
Supersizing Molecular Cloning by Rewriting the Rules for Golden Gate Assembly
Posted on Friday, June 11, 2021
By
Topic: What is Trending in Science
NEB scientists are getting help from DAD (Data-optimized Assembly Design) to construct HMW DNA targets
Cloning has come a long way since the first synthesis of recombinant DNA molecules using restriction enzymes in the 1970s. Now, variations on the traditional cut-and-paste workflow have led to the development of techniques such as Golden Gate Assembly (GGA), which enables directional assembly of multiple fragments in one tube. Recently, scientists at New England Biolabs (NEB) have assembled the entire 40 kb genome of the T7 bacteriophage from 52 fragments! In a new publication, NEB scientists describe how they rewrote the rules for assembly design and modified the traditional GGA protocols to rapidly construct large targets from many fragments.
In Golden Gate Assembly, the use of Type IIS restriction enzymes (REs) is a key feature that allows for multi-fragment assembly. Traditional cloning uses Type IIP REs that cut within their recognition sequence. In contrast, GGA uses Type IIS REs that cut outside of their recognition sequence – the significance of this is that it eliminates the recognition sequence from the final assembly product, allowing for digestion and ligation to occur in the same tube, and enabling seamless assembly. Not only does this simplify the experiment and reduce the hands-on time, it also gives the researcher the flexibility to choose the overhang sequences that join the fragments together – one of the most critical factors in assembly design as it determines capacity (how many fragments you can assemble) and fidelity (the ability to minimize misordered fragments) of the reaction.

Figure 1: Comparison of Type IIS and Type IIP cleavage sites with respect to their recognition sequences
If you’re new to GGA, you can check out this workflow video.
There are theoretical guidelines to consider when designing your overhangs:
(1) Palindromic sequence rule
(2) Duplicate sequence rule
These first two rules state that you want to avoid palindromes and duplicates – when present, you don’t have control of the ordering of the sequences.
(3) 3-base rule – avoid overhangs with the same 3 nucleotides in a row
(4) 2-base rule – avoid overhangs with 2 nucleotides in the same position
(5) Goldilocks rule – aptly named because you don’t want too much, or too little, GC content. You guessed it – GC content needs to be “just right.”
But, when you’re designing a large cloning system, sticking to all of these rules is difficult. Plus, you start to lose overhang sequence combinations, and you’re left with very little choice. These rules are based on general principles, not actual data, and so with this in mind, Pryor et al. set out to make assembly of large multi-fragment constructs easier and more robust.
This goal was approached from three angles:
(1) Optimize formulations of enzyme mixes for GGA;
(2) Develop a suite of webtools (see the end of this post for descriptions), and
(3) Utilize a data-driven approach to the selection of overhang sequences called Data-optimized Assembly Design, affectionately known as DAD. Pryor et al. did this by embarking on comprehensive profiling of overhang sequences to determine which ones work the best for adding flexibility and capacity to GGA. In effect, they rewrote the rules for assembly design. What they came up with allows for more flexibility when constructing complex, high molecular weight DNA targets for a wide range of synthetic biology applications.
Data-optimized Assembly Design maximizes GGA capacity
The goal here was to examine the fidelity and bias of every possible overhang sequence combination for use as a basis for overhang sequence selection. A next generation sequencing assay was developed whereby a hairpin substrate containing a Type IIS recognition sequence and randomized bases at the cleavage site was constructed. They conducted an assay for all of the most commonly used Type IIS restriction enzymes (BsaI-HF®v2, BsmBI-v2, Esp3I, BbsI) and even SapI (which leaves a 3 bp overhang sequence).
During incubation with a GG enzyme mix containing the Type IIS RE and T4 DNA Ligase, the substrate was cleaved to DNA intermediates with 4 bp overhang sequences (or 3 bp overhang sequence in the case of SapI). Within that intermediate pool existed representation of every possible sequence combination of a 4 (or 3) bp overhang. These DNA intermediates joined together to form a dumbbell assembly product that was sequenced using the Pacific BioSciences® (PacBio®) single-molecule sequencing platform.
The assay output was an enormous table with every possible sequence overhang combination and the number of reads for each overhang – in other words, a report of assembly bias. If a particular overhang was read more or less frequently, it was because this particular overhang was favored or disfavored, respectively, in the GG assembly.
Additional output included the frequency with which each overhang paired with its Watson-Crick partner, as well as the frequency and identity of mismatched partners – in other words, a report of assembly fidelity.
This data can be used as a tool to predict GGA success depending on the overhang sequences.
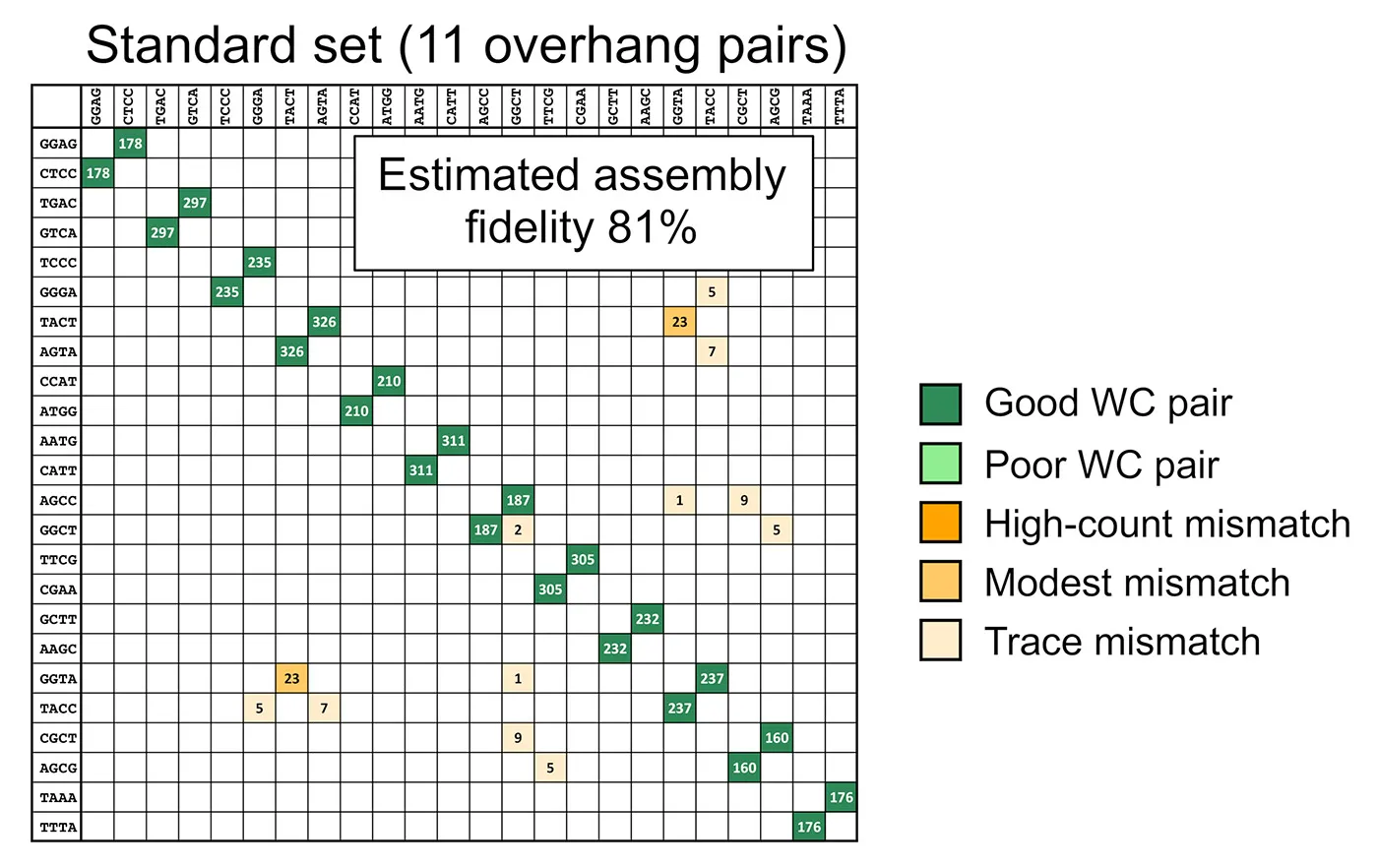
Figure 2: The data can be used as a predictive tool. The output is an estimation of assembly fidelity and the locations that mismatches are expected to occur.
Using DAD-based predictions, one can also estimate the maximum limits of GGA capacity – what is the highest fidelity you can identify given a certain number of overhang pairs in a reaction? So, in the graph below, you can see nearly perfect fidelity of up to about 20 overhang pairs.
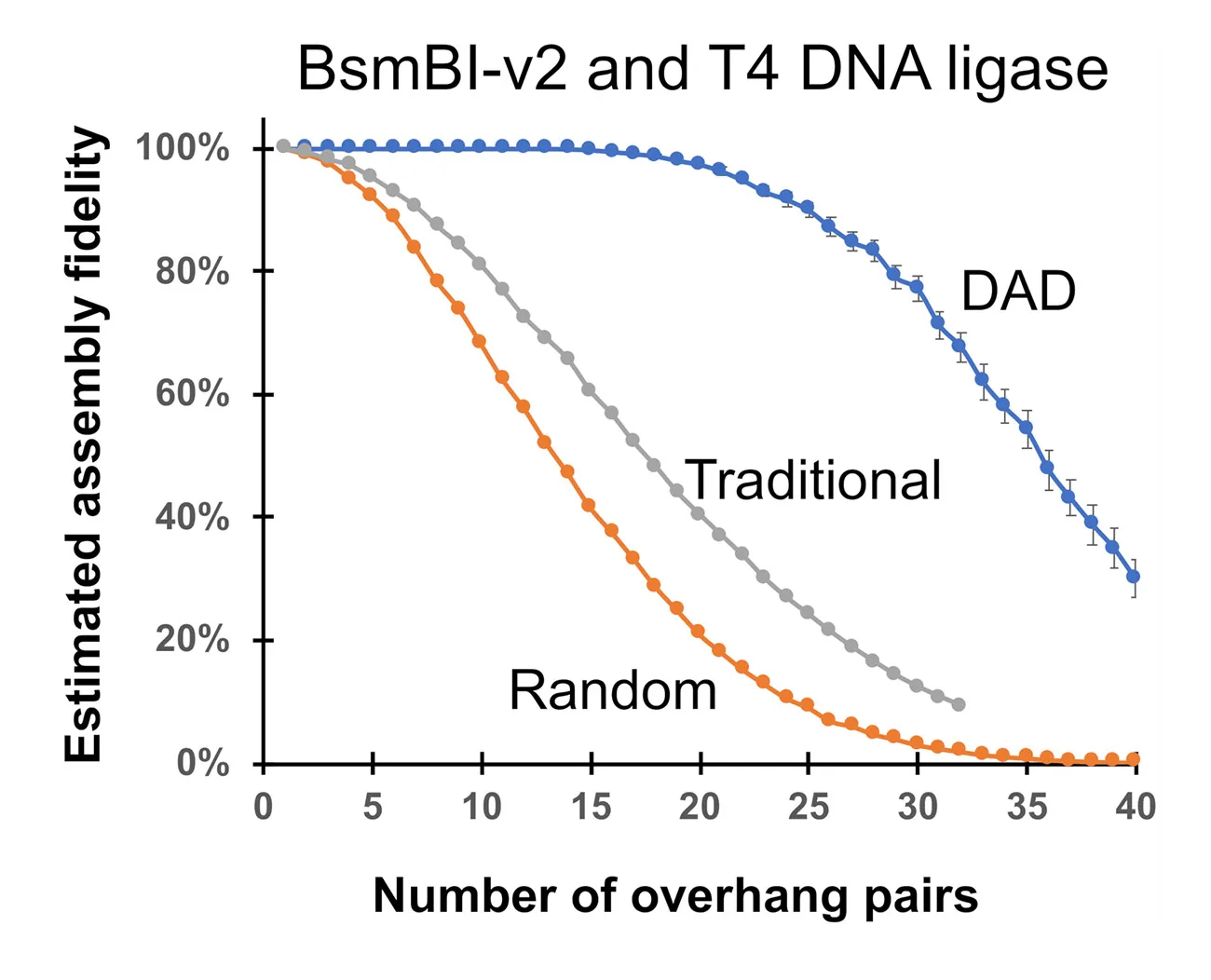
Figure 3: DAD suggests large assemblies are possible. When we compare Traditional rules for overhang selection (using theoretical guidelines #1-5) to Random (only using theoretical guidelines #1 & 2) to those selected by DAD, overhang selection using the theoretical guidelines gives just 5-10 overhang pairs with 100% fidelity, which is much less than the predictions based on DAD (20 overhang pairs).
Testing DAD predictions on a lac operon assembly
It should be noted though, that these are all just predictions. To put DAD to the test, Pryor et al. developed a reverse lac operon blue/white screen. Donor vectors each contained a small piece of the 5 kb lac operon cassette. When assembled into a destination vector, transformed into E. coli cells and plated onto an antibiotic plate with IPTG and X-gal, blue colonies formed if all the fragments were correctly assembled. They found that the predictions based on DAD calculations closely matched their experimental observations.
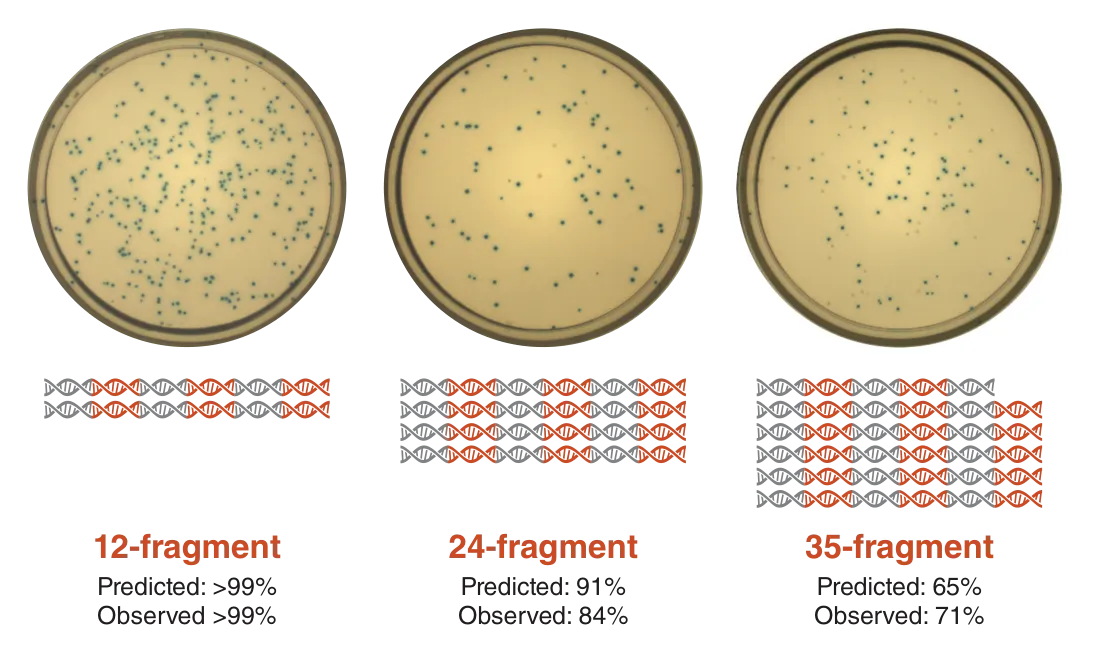
Figure 4: Predictions based on DAD calculations matched the experimental outcomes
The effect of a static incubation temperature
To push the fragment limit even further, the next step was to modify experimental conditions with regard to temperature and cycling. Instead of using oscillating temperatures typically used in GGA, a static 37°C temperature was used. This temperature resulted in a 2-fold reduction of the frequency of mismatches. And, while 37°C isn’t the ideal temperature for ligation, the decrease in ligation efficiency could be offset by a longer incubation period. So, a higher static 37°C temperature and an extended incubation resulted in increased fidelity and almost equivalent efficiency. The benefit of the static 37°C incubation temperature over a 37/16°C oscillating cycle was observed once the assembly complexity reached 35 fragments and greater.
After pushing the lac operon assembly capacity to 52 fragments (not pictured), using DAD and a static 37°C incubation, Pryor et al. decided to assemble a much higher molecular weight target than the 5 kb lac operon: the 40 kb T7 bacteriophage.
Using their cloning expertise and DAD to construct the 40kb T7 bacteriophage genome
Using one of our GGA webtools, NEBridge SplitSet® (see below), Pryor et al., generated primers for 52 DNA fragments representing the T7 bacteriophage genome. NEBridge SplitSet is a great tool for those new to GGA because you can simply upload your sequence and receive output that will give you an indication of how your assembly will look. With the ability to specify search windows, the biology can determine the breakpoints, and you can line up the junctions at the boundaries of open-reading frames.
Using GGA, the bacteriophage genome was assembled using a static 37°C temperature and transformed into E. coli, which successfully reconstituted the infectious phage. They verified the assembly using plaque PCR plus or minus restriction digest.
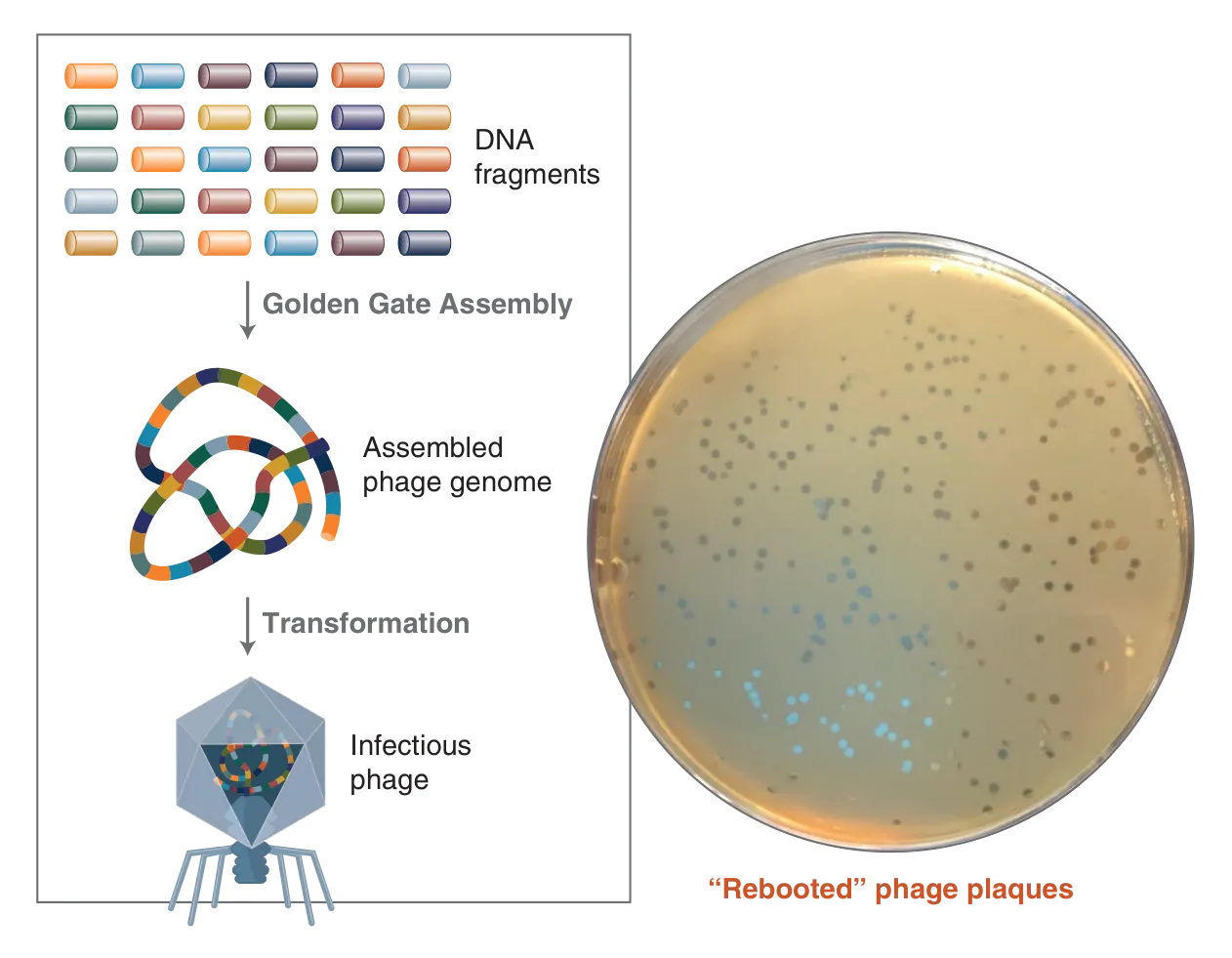
Figure 5: 52-part T7 phage genome assembly workflow
The field of synthetic biology is expanding at an exciting speed that is hard to keep up with! The rapid and robust construction of large DNA targets, such as small genomes or even entire metabolic pathways, means that time is not wasted doing multiple hierarchical assembly rounds - researchers can get to their downstream experiments sooner. The beauty of Golden Gate Assembly is that fragments can be sourced from libraries of predefined DNA fragments and assembled in a single step, in a single tube. As an added bonus, GGA is amenable to automation! NEB researchers have shown that DAD, when applied to GGA, can robustly and reliably facilitate the assembly of >50 fragments in a single reaction – a big step in supersizing high molecular weight complex DNA targets.
Check out our web tools created by NEB researchers - they will help you guide your assembly design, regardless of the number of fragments, with less guesswork.
Here are some of our webtools that can make your assembly design easier
NEBridge Ligase Fidelity Viewer® - as the name suggests, this tool estimates assembly fidelity. It provides you with a mismatch matrix so you can identify the overhangs that are the most prone to mismatch.
NEBridge GetSet® – with this tool, you can specify parameters such as the number of overhang sets you want to assemble and overhangs that you require to be included or excluded. The output is an estimated assembly fidelity, the mismatch matrix, and the overhang sequences.
NEBridge SplitSet – no need to concern yourself with overhangs using this tool. You just upload the sequence that you want to assemble. Specify the number of fragments and even the position and approximate search windows for breakpoints in your sequence. The output is the estimated assembly fidelity, mismatch matrix for the overhangs, the sequences for the component fragments and the PCR primers for generation of the assembly parts.
Read the full story of how NEB researchers enabled the one-pot assembly of large complex DNA targets using DAD, and how they used DAD to rapidly construct the 40 kb genome T7 bacteriophage genome.
You can also check out Listen to DAD when constructing high-complexity Golden Gate Assembly targets from the NEB TV Webinar series.
NEB will not rent, sell, or otherwise transfer your data to a third party for monetary consideration. See our Privacy Policy for details. View our Community Guidelines.
Products and content are covered by one or more patents, trademarks and/or copyrights owned or controlled by New England Biolabs, Inc (NEB). The use of trademark symbols does not necessarily indicate that the name is trademarked in the country where it is being read; it indicates where the content was originally developed. All other trademarks are the property of their respective owners. The use of this product may require the buyer to obtain additional third-party intellectual property rights for certain applications. For more information, please email busdev@neb.com.
Don’t miss out on our latest NEBinspired blog releases!
- Sign up to receive our e-newsletter
- Download your favorite feed reader and subscribe to our RSS feed
Be a part of NEBinspired! Submit your idea to have it featured in our blog.

