Anatomy of a Polymerase - How Function and Structure are Related
Accurate genome replication is critical for the viability of any organism. The general concept for copying DNA was evident upon the elucidation of DNA’s double helical structure and the identification of base pair complementarity (1): one strand of nucleobases could serve as the template for synthesis of a new strand. Within a decade of these discoveries, an agent was purified from E. coli that catalyzed DNA strand duplication (2). This agent was termed a "polymerase". E. coli DNA Polymerase I, the first DNA polymerase discovered was not the primary replicative polymerase, but instead one involved in lagging strand Okazaki fragment resolution and DNA repair. This foreshadowed future discoveries of many DNA polymerase families, each serving specific cellular requirements for DNA replication and repair.
DNA Polymerases serve as fundamental enzymes in the life sciences for the same reason that they are critical in nature: they copy DNA. Polymerase applications include DNA labeling, sequencing and amplification. One specific amplification protocol, the polymerase chain reaction (PCR) is a widely used technique that employs thermophilic polymerases to exponentially amplify specific DNA segments (3) and enables a range of applications from human and pathogen diagnostics to molecular cloning in biology labs around the world.
Polymerase Accuracy
PCR puts the same basic demands on a polymerase as a cell puts on its replication system. Essentially,the polymerase should be reliable, accurate and fast. Polymerase accuracy or “fidelity” refers to the propensity to incorporate the correct nucleotide as specified by the template strand. The standard PCR enzymes are, not surprisingly, quite accurate. Even Thermus aquaticus (Taq) DNA polymerase, considered a low fidelity PCR polymerase, only makes a mistake once in approximately 6,000 nucleotide insertions (12). Polymerase discovery and engineering efforts have produced high fidelity polymerases, which rarely make base substitution mistakes, requiring DNA sequencing methods to read millions of synthesized bases to detect any errors by the polymerase Advancements in measuring fidelity by single-molecule sequencing has identified Q5® High-Fidelity DNA Polymerase as having fidelity 280X greater thanTaq DNA polymerase (12).
Fidelity checkpoints: geometric selection at the active site and beyond
DNA polymerases ensure accurate replication using a series of molecular checkpoints, at the site of nucleotide incorporation and beyond (1). During nucleotide addition, the correct incoming nucleotide is positioned for a productive alignment of catalytic groups, ensuring efficient incorporation. This alignment for catalysis is sensitive to distortions in position caused by incorrect Watson-Crick base pairing, allowing for kinetic stalling at incorrect or non-cognate base pairs.
Proofreading of DNA Polymerases
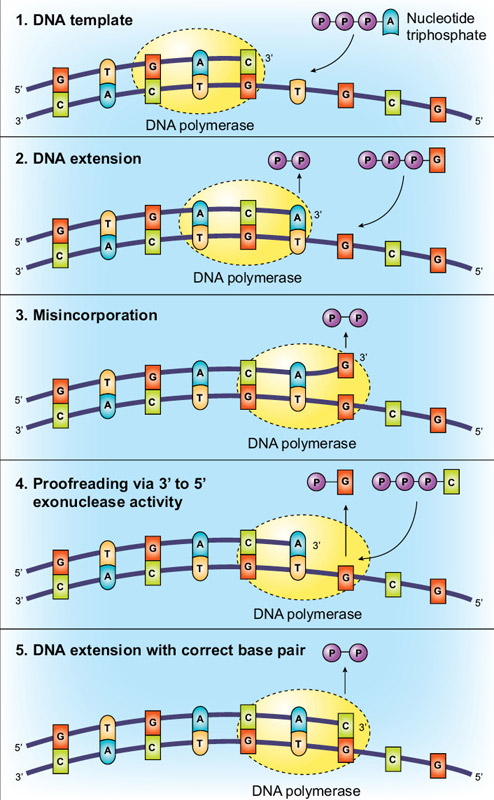
Another method of increasing fidelity is for the polymerase to have 3´→5´ exonuclease activity, termed “proofreading”. Using the base-pairing and active site molecular checkpoints described above, Taq DNA polymerase is incredibly accurate, but proofreading enzymes can have even higher fidelity. This additional accuracy is conveyed via proofreading, with the polymerase “checking” whether the correct nucleotide has been inserted into the template. If a mismatch is detected the DNA is transferred from the polymerization domain to an N-terminal 3´→5´ exonuclease domain of the polymerase. The incorrectly incorporated nucleotide is excised,DNA moves back into the polymerization domain, and copying can resume (Figure 2).
Bacteriophage T4 proved to be a useful experimental system for evaluating the importance of 3´→5´ exonuclease activity for accurate DNA replication (6). Mutations in T4 gene 43 (which encodes the DNA polymerase) were identified that either decreased or increased fidelity. By defining an exonuclease/polymerase (N/P) activity ratio for a mutant enzyme it was found that polymerases with low N/P ratios were more error prone than those with high N/P ratios. An explanation for this observation is that upon incorporation of a mismatched base it is more likely that the exonuclease will remove the nucleotide before the polymerase activity extends it in enzymes with higher N/P ratios. Interestingly, the proofreading effectiveness of a polymerase can show sequence dependence. For example, AT-rich sequences are more effectively proofread than GC regions. This discrepancy is thought to be due to the lower stability of AT regions that facilitates strand melting and therefore, proofreading activity.
The absence of 3´→5´ exonuclease activity may have ramifications other than fidelity in PCR. The lack of proofreading activity in Taq DNA Polymerase has been proposed to limit the amplicon size possible with this enzyme (7). Generally, Taq performs best when amplifying DNA fragments < 2 kb, and can work with fragments up to 3–4 kb. When kept to this amplicon size, Taq is a robust, easily optimized enzyme. However, above ~3 kb it quickly drops in effectiveness. During PCR, Taq will misincorporate nucleotides and produce mismatches, at which it is prone to stalling and is more likely to dissociate before extending as compared to correctly base paired 3´ ends. Therefore, at a certain amplicon size and polymerase error rate enough mismatched 3´ ends may accumulate to effectively inhibit the PCR process. These mismatched 3´ ends are particularly problematic for Taq because it lacks the 3´→5´ exonuclease activity to remove them. By adding in a small amount of proofreading enzyme such as Deep Vent®DNA Polymerase, amplification of fragments ≥ 20 kb can be achieved (Figure 3). Since the vast majority of the enzyme in the blend is Taq DNA Polymerase it is probably doing the bulk of the primer extension, with the proofreading Deep Vent polymerase removing the inhibitory 3´ mismatches generated by Taq.

Polymerase Processivity
The importance of proofreading activity to PCR has been widely known for nearly two decades, but another property, processivity, has gained increased attention. “Processivity” is a term that refers to the number of nucleotides incorporated by a polymerase in a single binding event (before dissociation). Taq DNA polymerase adds approximately 50 nucleotides per binding event (8). Why does this matter? A low-processivity or “distributive” polymerase extends a population of templates in a noticeably different manner than a processive polymerase. A highly distributive polymerase binds to a template, adds a couple of nucleotides, and dissociates, leaving a population of templates that can be extended equally with time. A highly processive polymerase binds a template and extends with longer binding events.
It would follow that given enough time the outcome of either a processive or distributive polymerase reaction would be a population of copied templates. However, in certain circumstances it is possible that the processive polymerase has superior performance. The E. coli polymerase III α subunit, part of the main replicative polymerase, has a processivity of < 10 base pairs and a speed of < 20 nucleotides/second (nt/s). However, when the subunit associates with the other replisome subunits, particularly the sliding clamp, the effective processivity and replication speed increase to > 50 kb and 1,000 nt/s, respectively (9). The term “effective processivity” is used because there is data indicating the polymerase subunit can exchange in the replisome, but the replisome maintains fast, processive DNA replication (10).
To take advantage of processivity in PCR, researchers have fused a DNA binding domain to an archaeal polymerase (11). This chimeric enzyme has several improved properties, but notably it is able to amplify DNA with shorter extension times and produce longer DNA products more efficiently, thus shortening overall thermocycling times. This fusion idea is the basis of Q5 High-Fidelity DNA Polymerase and Phusion® High-Fidelity DNA Polymerase, two polymerases available from NEB (Figure 4).
Future Directions
Many properties affect the efficacy and utility of a PCR polymerase. Polymerase active site architecture and proofreading activity affect the accuracy of the final product. Polymerase blends and fusion to a DNA binding protein confer superior PCR performance for amplicon length and, in the case of the chimera, reaction speed. Other important advances in PCR, such as hot-start polymerases to increase reaction specificity, multiplex PCR (Figure 5) and qPCR have also revolutionized the life sciences.
As demonstrated by engineered blends and chimeras, properties of the polymerase itself can be modulated to improve PCR performance. In the future, it is likely that polymerase properties will increasingly be tailored to specific PCR applications, and as such, this is an important area of research at NEB.

View NEB’s DNA Polymerase Selection Chart
References
- Watson, J. D. & Crick, F. H. (1953) Nature, 171, 964–967
- Lehman, I. R. (2003) J. Biol. Chem. 278, 34733–34738
- Saiki, R. K., et al. (1988) Science, 239, 487–491
- Eckert, K. A. & Kunkel, T. A. (1990) Nucleic Acids Res. 18, 3739
- Liu, D., Moran, S., & Kool, E. T. (1997) Chem. Biol. 4, 919–926
- Goodman, M. F. & Fygenson, D. K. (1998) Genetics, 148, 1475–1482
- Barnes, W. M. (1994) Proc. Natl. Acad. Sci. USA, 91, 2216–2220
- Merkens, L. S., Bryan, S. K., & Moses, R. E. (1995) Biochim Biophys. Acta, 1264, 243-248
- Pomerantz, R. T. & O’Donnell, M. (2007) Trends Microbiol. 15, 156–164
- Lovett, S. T. (2007) Mol. Cell, 27, 523–526
- Wang, Y., et al. (2004) Nucleic Acids Res. 32, 1197–11207
- Potapov, V. and Ong, J.L. (2017) PLoS ONE. 12(1): e0169774).
