Restriction Endonucleases: Molecular Cloning and Beyond
Siu-Hong Chan, Ph.D., New England Biolabs, Inc.
INTRODUCTION
In the 1950s, a phenomenon known as “host controlled/induced variation of bacterial viruses” was reported, in which bacteriophages isolated from one E. coli strain showed a decrease in their ability to reproduce in a different strain, but regained the ability in subsequent infection cycles (1,2). In 1965, Werner Arber’s seminal paper established the theoretical framework of the restriction-modification system, functioning as bacterial defense against invading bacteriophage (3). The first REases discovered recognized specific DNA sequences, but cut at variable distances away from their recognition sequence (Type I) and, thus were of little use in DNA manipulation. Soon after, the discovery and purification of REases that recognized and cut at specific sites (Type II REases) allowed scientists to perform precise manipulations of DNA in vitro, such as the cloning of exogenous genes and creation of efficient cloning vectors. Now, more than 4,000 REases are known, recognizing more than 300 distinct sequences (for a full list, visit REBASE® at rebase.neb.com). With the advent of the Polymerase Chain Reaction (PCR), RT-PCR, and PCR-based mutagenesis methodologies, the traditional cloning workflow transformed biological research in the decades that followed.

ENGINEERING OF RESTRICTION ENZYMES
Traditionally, REases were purified from the native organism. The development of gene cloning vectors and selection methodologies enabled the cloning of REases. Cloning not only allowed the production of large quantities of highly purified enzymes, but also made the engineering of REases possible. Currently, > 250 of the restriction enzymes supplied by New England Biolabs (NEB) are recombinant proteins.
Engineering Improved Performance
Cleavage activity at non-cognate sites (i.e., star activity) had been observed and well-documented for some REases. Of those, some exhibit star activity under sub-optimal reaction conditions, while others have a very narrow range of enzyme units that completely digest a given amount of substrate without exhibiting star activity (4). Through intensive research, scientists at NEB began engineering restriction enzymes that exhibit minimal, if any, star activity with extended reaction times and at high enzyme concentrations. This research enabled the introduction of High Fidelity (HF™) REases that have improved performance under a wider range of reaction conditions (for more information, visit www.neb.com/HF).
Engineering New Sequence Specificities
Attempts to alter the sequence specificities of Type IIP REases have been largely unsuccessful, presumably because the sequence specificity determinant is structurally integrated with the active sites of Type IIP REases. MmeI, a Type IIG REase with both methyltransferase (MTase) and REase activities in the same polypeptide, recognizes the target sequence TCCRAC using the target recognition domain (TRD) within its MTase component. This represented an excellent opportunity to engineer altered sequence specificity into the REase. As an added advantage, the sharing of the TRD between the REase and MTase activities resulted in an equivalent change in MTase activity for any change in target sequence cleavage specificity, protecting the new target site from cleavage in recombinant host cells. Through bioinformatics analysis of homologous protein sequences, scientists at NEB identified the amino acid residues that recognized specific bases within the target sequences and created MmeI mutants with altered sequence specificities (5). Rational design of MmeI mutants and homologs unlocked the potential for the creation of REases with hundreds of new sequence specificities.
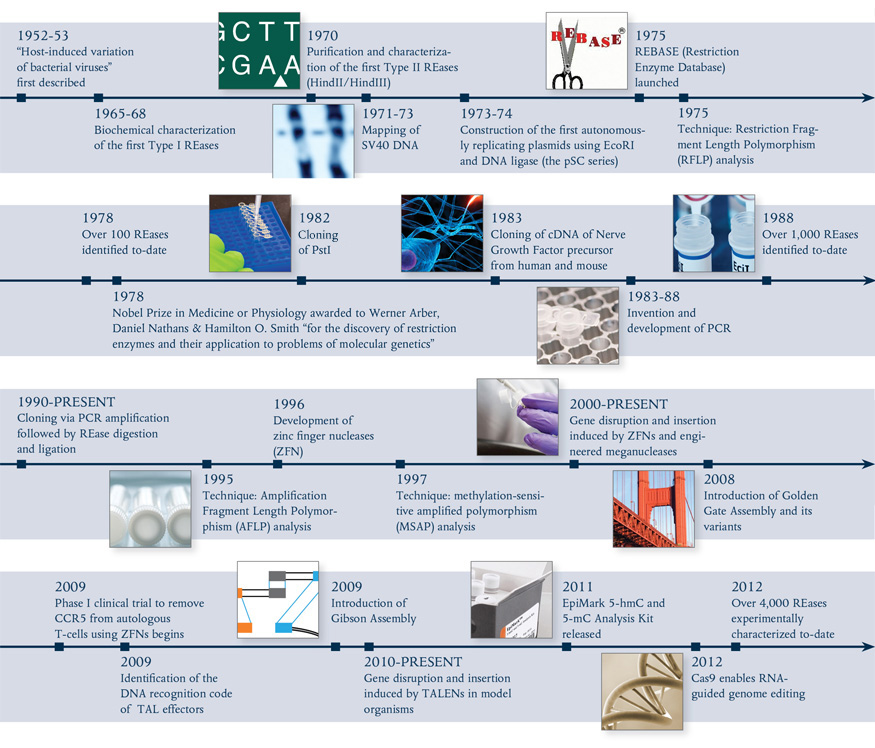
Type IIS REases, such as FokI (light and dark brown) and BstNBI (isoschizomer of BspD6I, light and dark purple), and homing endonuclease I-AniI (cyan), have been engineered to posses nicking enzyme activities.
Engineering Nicking Endonucleases
Basic research involving REases led to surprising findings about the seemingly straightforward mechanism of cleavage. Prototypical Type IIP REases normally act as homodimers, with each of the monomers nicking half of the palindromic site. Type IIS REases, on the other hand, exhibit a broad range of double-stranded cleavage mechanisms, namely heterodimerization, as by BtsI and BbvCI, and sequential cleavage of the dsDNA as monomer, as by FokI. These properties have been exploited to create strand-specific nicking enzymes (NEases) (for more information about nicking enzymes, see review in (6)).
APPLICATIONS UTILIZING RESTRICTION ENZYMES
Traditional Cloning
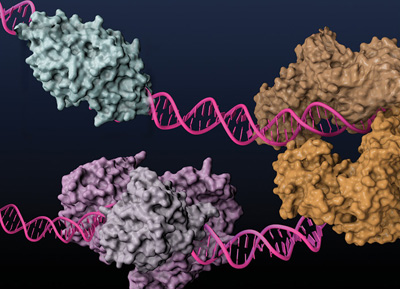
In combination with DNA ligases, REases facilitated a robust “cut and paste” workflow where a defined DNA fragment could be moved from one organism to another (Fig. 2). Using this methodology, Stanley Cohen and his colleagues incorporated exogenous DNA into natural plasmids to create the vehicle for cloning-plasmid vectors that self-propagate in E. coli (7). These became the backbone of many present-day vectors, and enabled the cloning of DNA for the study and production of recombinant proteins. Restriction enzymes are also useful as post-cloning confirmatory tools, to ensure that insertions have taken place correctly. The traditional cloning workflow, along with DNA amplification technologies, such as PCR and RT-PCR, has become a mainstream application for REases and facilitated the study of many molecular mechanisms.
Using PCR, restriction sites are added to both ends of a dsDNA, which is then digested by the corresponding REases. The cleaved DNA can then be ligated to a plasmid vector cleaved by the same or compatible REases with T4 DNA ligase. DNA fragments can also be moved from one vector into another by digesting with REases and ligating to compatible ends of the target vector.
DNA Mapping
Armed with only a handful of REases in the early 1970s, Daniel Nathans mapped the functional units of SV40 DNA (8), and commenced the era of “restriction mapping” and comparison of complex genomes. It has since evolved into sophisticated methodologies that allow the detection of single nucleotide polymorphisms (SNP) and insertions/deletions (Indels) (9), driving applications that include identifying genetic disorder loci, assessing the genetic diversity of populations and parental testing.
Understanding Epigenetic Modifications
REases’ sensitivity to the methylation status of target bases has been exploited to map modified bases within genomes. Restriction Landmark Genome Scanning (RLGS) is a 2-dimensional gel electrophoresis-based mapping technique that employs NotI (GC^GGCCGC), AscI (GG^CGCGCC), EagI (C^GGCCG) or BssHII (G^CGCGC) to interrogate changes in the methylation patterns of the genome during the development of normal and cancer cells. Methylation-Sensitive Amplification Polymorphism (MSAP) takes advantage of the differential sensitivity of MspI and HpaII toward the methylation status of the second C of quadruplet CCGG to identify 5-methylcytosine (5-mC) or 5-hydroxymethylcytosine (5-hmC) (10,11). Scientists at NEB further exploited the property of MspI and HpaII on 5-glucosyl hydroxymethylcytosine (5-ghmC) in the EpiMark® 5-hmC and 5-mC Analysis Kit (NEB #E3317S)(12), which differentiates 5-hmC from 5-mC for more refined epigenetic marker identification and quantitation (for more information, visit EpiMark.com). Additionally, the recently discovered REases that recognize and cleave DNA at 5-mC and 5-hmC sites (e.g., MspJI, FspEI and LpnPI), as well as those that preferentially cleave 5-hmC or 5-ghmC over 5-mC or C (e.g., PvuRts1I, AbaSI) (13), are potential tools for high-throughput mapping of the cytosine-based epigenetic markers in cytosine-methylated genomes (14,15).
In vitro DNA Assembly Technologies
Synthetic biology is a rapidly growing field, in which defined components are used to create biological systems for the study of biological processes and the creation of useful biological devices (16). Novel technologies such as BioBrick™ originally emerged to facilitate the building of such biological systems. Recently, more robust approaches, such as Golden Gate Assembly and Gibson Assembly™, have been widely adopted by the synthetic biology community. Both approaches allow for the parallel and seamless assembly of multiple DNA fragments without resorting to non-standard bases.
BioBrick: The BioBricks community sought to create thousands of “standardized parts” of DNAs for rapid gene assembly. With the annual International Genetically Engineered Machines (iGEM) competition (igem.org), the BioBricks community grew and elicited broad interest from many university students in synthetic biology. Based on traditional REase-ligation methodology, BioBrick and its derivative methodologies (BioBrick Assembly Kit, NEB #E0546, and its derivative, BglBricks (17)) are easy to use, but they introduce scar sequences at the junctions. They also require multiple cloning cycles to create a working biological system.
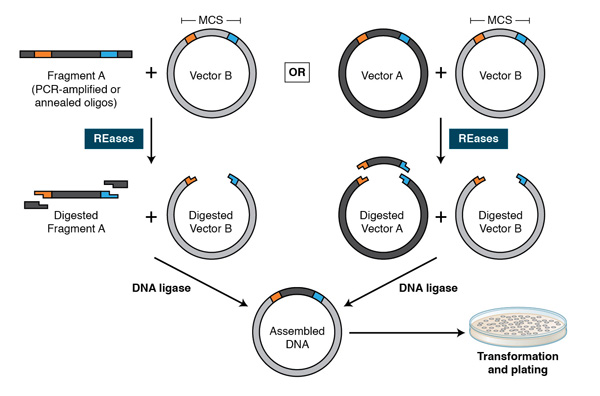
Golden Gate Assembly: Golden Gate Assembly and its derivative methods (19,20) exploit the ability of Type IIS REases to cleave DNA outside of the recognition sequence. The inserts and cloning vectors are designed to place the Type IIS recognition site distal to the cleavage site, such that the Type IIS REase can remove the recognition sequence from the assembly (Fig. 3). The advantages of such an arrangement are three-fold: 1. the overhang sequence created is not dictated by the REase, and therefore no scar sequence is introduced; 2. the fragment-specific sequence of the overhangs allows orderly assembly of multiple fragments simultaneously; and 3. the restriction site is eliminated from the ligated product, so digestion and ligation can be carried out simultaneously. The net result is the ordered and seamless assembly of DNA fragments in one reaction. The accuracy of the assembly is dependent on the length of the overhang sequences. Therefore, Type IIS REases that create 4-base overhangs (such as BsaI/BsaI-HF, BbsI, BsmBI and Esp3I) are preferred. The downside of these Type IIS REase-based methods is that the small number of overhanging bases can lead to the mis-ligation of fragments with similar overhang sequences (21). It is also necessary to verify that the Type IIS REase sites used are not present in the fragments for the assembly of the expected product. Nonetheless, Golden Gate Assembly is a robust technology that generates multiple site-directed mutations (22) and assembles multiple DNA fragments (23,24). As open source methods and reagents have become increasingly available (see www.addgene.org), Golden Gate Assembly has been widely used in the construction of custom-specific TALENs for in vivo gene editing (25), among other applications.
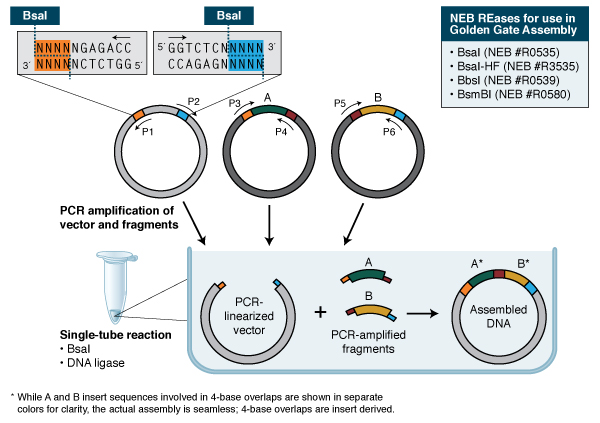
In its simplest form, Golden Gate Assembly requires a BsaI recognition site (GGTCTC) added to both ends of a dsDNA fragment distal to the cleavage site, such that the BsaI site is eliminated by digestion with BsaI or BsaI-HF (GGTCTC 1/5). Upon cleavage, the overhanging sequences of the adjoining fragments anneal to each other. DNA ligase then seals the nicks to create a new covalently linked DNA molecule. Multiple pieces of DNA can be cleaved and ligated simultaneously.
Gibson Assembly can also be used for cloning; the assembly of a DNA insert with a restriction-digested vector, followed by transformation, can be completed in a little less than two hours with the Gibson Assembly Cloning Kit (NEB #E5510S, for more information, visit NEBGibson.com). Other applications of Gibson Assembly include the introduction of multiple mutations, assembly of plasmid vectors from chemically synthesized oligonucleotides, and creating combinatorial libraries of genes and pathways.
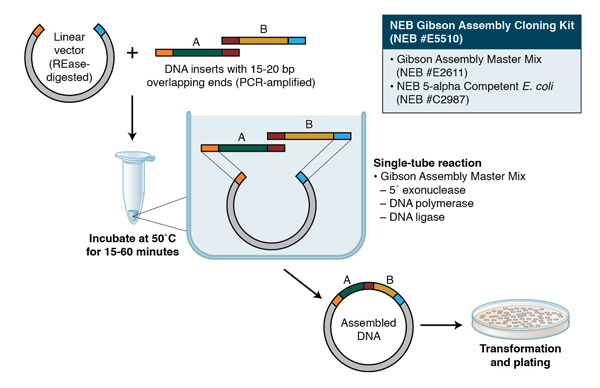
Gibson Assembly employs three enzymatic activities in a single-tube reaction: 5´ exonuclease, the 3´ extension activity of a DNA polymerase and DNA ligase activity. The 5´ exonuclease activity chews back the 5´ end sequences and exposes the complementary sequence for annealing. The polymerase activity then fills in the gaps on the annealed regions. A DNA ligase then seals the nick and covalently links the DNA fragments together. The overlapping sequence of adjoining fragments is much longer than those used in Golden Gate Assembly, and therefore results in a higher percentage of correct assemblies. The NEB Gibson Assembly Master Mix (NEB #E2611) and Gibson Assembly Cloning Kit (NEB #E5510S) enable rapid assembly at 50˚C.
Construction of DNA Libraries
SAGE (Serial Analysis of Gene Expression) has allowed the identification and quantification of a large number of mRNA transcripts. It has been widely used in cancer research to identify mutations and study gene expression. REases are key to the SAGE workflow. NlaIII is instrumental as an anchoring enzyme, because of its unique property of recognizing a 4-bp sequence CATG and creating a 4 nucleotide overhang of the same sequence. The use of Type IIS enzymes as tagging enzymes that cleave further and further away from the recognition sequence allows for the higher information content of SAGE analyses (e.g., FokI and BsmFI in SAGE (28), MmeI in LongSAGE (29) and EcoP15I in SuperSAGE (30) and DeepSAGE (31)).
Chromosome conformation capture (3C) and derivative methods allow the mapping of the spatial organizations of genomes in unprecedentedly high resolution and throughput (32). REases plays an indispensible role in creating the compatible ends of the DNA cross-linked to its interacting proteins, such that spatially associated sequences can be ligated and, hence, identified through high-throughput sequencing.
Although REases do not allow for the random fragmentation of DNA that most next-generation DNA sequencing technologies require, they are being used in novel target enrichment methodologies (hairpin adaptor ligation (33) and HaloPlex™ enrichment (Agilent)). The long-reach REase, AcuI, and USER™ Enzyme are also used to insert tags into sample DNA, which is then amplified by rolling circle amplification (RCA) to form long, single-stranded DNA “nanoballs” that serve as template in the high density, chip-based sequencing-by-ligation methodology, developed by Complete Genomics (34). ApeKI was also used to generate the DNA library for a genotyping-by-sequencing technology for the study of sequence diversity of maize (35).
Creation of Nicks in DNA
Before NEases were available, non-hydrolyzable phosphorothioate groups were incorporated into a specific strand of the target DNA such that REases can introduce sequence- and strand-specific nicks into the DNA for applications such as strand displacement amplification (SDA), where a strand-displacing DNA polymerase (e.g., Bst 2.0 DNA Polymerase, NEB #M0537) extends from the newly created 3’-hydroxyl end, and essentially replicates the complementary sequence (36). Because the nicking site is regenerated, repeated nicking-extension cycles result in amplification of specific single-stranded segments of the sample DNA without the need for thermocycling. NEases greatly streamline the workflow of such applications and open the door to applications that cannot be achieved by REases. Nicking enzyme-based isothermal DNA amplification technologies, such as RCA, NESA, EXPAR and related amplification schemes, have been shown to be capable of detecting very low levels of DNA (37,38). Nicking-based DNA amplification had also been incorporated into molecular beacon technologies to amplify signal (39). The implementation of these sample and/or signal amplification schemes can lead to simple, but sensitive and specific, methods for the detection of target DNA molecules in the field (NEAR, EnviroLogix™). By ligating adaptors containing nicking sites to the ends of blunt-ended DNA, the simultaneous actions of the NEase(s) and strand-displacing DNA polymerase can quickly amplify a specific fragment of dsDNA (40). Amplification by nicking-extension cycling is amenable to multiplexing and can potentially achieve a higher fidelity than PCR. The combined activity of NEases and Bst DNA polymerase have also been used to introduce site-specific fluorescent labels into long/chromosomal DNA in vitro for visualization (nanocoding) (41). Innovative applications of nicking enzymes include the generation of reporter plasmids with modified bases or structures (42) and the creation of a DNA motor that transports a DNA cargo without added energy (43). A review of NEases and their applications has been published elsewhere (6).
In vivo Gene Editing
The ability to “cut and paste” DNA using REases in vitro has naturally led to the quest for performing the art in vivo to correct mutations that cause genetic diseases. Direct use of REases and homing endonucleases in Restriction Enzyme Mediated Integration (REMI) facilitated the generation of transgenic embryos of higher organisms (44,45). There is, however, no control over the integration site. The concept of editing genes through site-specific cleavage has been realized using Zinc Finger Nucleases (ZFNs) and Transcription Activator-like Effector Nucleases (TALENs), due to their ability to create customizable double stranded breaks in complex genomes. With the great success of gene editing in model organisms and livestock (46-50), the therapeutic potential of these gene editing reagents is being put to the first test in the Phase I/II clinical trials of a regime that uses a ZFN to improve CD4+ T-cell counts by knocking out the expression of the CCR5 gene in autologous T-cells from HIV patients (ClinicalTrials.gov indentifier NCT00842634) (51). Recent research on CRISPR, the adaptive defense system of bacteria and archaea, has shown the potential of the Cas9-crRNA complex as programmable RNA-guided DNA endonucleases and strand-specific nicking endonucleases for in vivo gene editing (52,53).
MOVING FORWARD
Restriction enzymes have been one of the major forces that enabled the cloning of genes and transformed molecular biology. Novel technologies, such as Golden Gate Assembly and Gibson Assembly, continue to emerge and expand our ability to create new DNA molecules. The potential to generate new recognition specificity in the MmeI family REases, the engineering of more NEases and the discovery of ever more modification-specific REases continues to create new tools for DNA manipulation and epigenome analysis. Innovative applications of these enzymes will take REases’ role beyond molecular cloning by continuing to accelerate the development of biotechnology and presenting us with new opportunities and challenges.
References- Bertani, G., and Weigle, J. (1953) J. Bact. 65, 113-121.
- Luria, S., and Human, M. (1952) J. Bact. 64, 557-569.
- Arber, W. (1965) Ann. Rev. Microbiol. 19, 365-378.
- Wei, H., et al. (2008) Nucl. Acids Res. 36, e50.
- Morgan, R. D., and Luyten, Y. A. (2009) Nucl. Acids Res. 37, 5222-5233.
- Chan, S., Stoddard, B. L., and Xu, S. (2011) Nuc.Acids Res. 39, 1-18.
- Cohen, S., et al. (1973) PNAS 70, 3240-3244.
- Danna, K., and Nathans, D. (1972) PNAS 69, 3097-3100.
- Kudva, I. T., et al. (2004) J. Clin. Microbiol. 42, 2388-2397.
- Reyna-Lopez, G., and Simpson, J. (1997) Mol. Gen. Genet. 253, 703-710.
- Mastan, S. G., et al. (2012) Gene 508, 125-129.
- Davis, T., and Vaisvila, R. (2011) J. Vis. Exp.: JoVE 2661.
- Zhu, Z., et al. (2011) Compositions, Methods And Related Uses For Cleaving Modified DNA, WO Patent WO 2011/091146 A1.
- Wang, H., et al. (2011) Nucl. Acids Res. 39, 9294-9305.
- Cohen-Karni, D., et al. (2011) PNAS 108, 11040-11045.
- Ellis, T., Adie, T., and Baldwin, G. S. (2011) Integr. Biol. (Camb) 3, 109.
- Anderson, J. C., et al. (2010) J. Biol. Eng. 4, 1-12.
- Nour-Eldin, H. H., Geu-Flores, F., and Halkier, B. A. (2010) Meth. Mol. Biol. 643, 185-200.
- Engler, C., Kandzia, R., and Marillonnet, S. (2008) PLoS ONE 3, e3647.
- Sarrion-Perdigones, A., et al. (2011) PLoS ONE 6, e21622.
- Engler, C., et al. (2009) PLoS ONE 4, e5553.
- Yan, P., et al. (2012) Anal. Biochem. 430, 65-67.
- Scior, A., et al. (2011) BMC Biotechnol. 11, 87.
- Werner, S., et al. (2012) Bioeng. Bugs 3, 38-43.
- Sanjana, N. E., et al. (2012) Nat. Protoc. 7, 171-192.
- Gibson, D. G., et al. (2009) Nat. Methods 6, 343-345.
- Gibson, D. G., et al. (2010) Science 329, 52-56.
- Velculescu, V. E., et al. (1995) Science 270, 484-487.
- Høgh, A. L., and Nielsen, K. L. (2008) Meth. Mol. Biol. 387, 3-24.
- Matsumura, H., et al. (2008) Meth. Mol. Biol. 387, 55-70.
- Nielsen, K. L. (2008) Meth. Mol. Biol. 387, 81-94.
- deWit, E. and deLaat, W. (2012) Genes Dev. 26, 11-24.
- Singh, P., Nayak, R., and Kwon, Y. M. (2011) Meth. Mol. Biol. 733, 267-278.
- Drmanac, R., et al. (2010) Science 327, 78-81.
- Elshire, R. J., et al. (2011) PLoS ONE 6, e19379.
- Spargo, C. A., et al. (1996) Mol. Cell Probes 10, 247-56.
- Dawson, E. D., et al. (2009) Mol. Biotech. 42, 117-127.
- Murakami, T., Sumaoka, J., and Komiyama, M. (2009) Nuc. Acids Res. 37, e19.
- Li, J. J., et al. (2008) Nucl. Acids Res. 36, e36.
- Joneja, A., and Huang, X. (2011) Anal. Biochem. 414, 58-69.
- Zhang, P., et al. (2010) Protein Expr. Purif. 69, 226-234.
- Luhnsdorf, B., et al. (2012) Anal. Biochem. 425, 47-53.
- Bath, J., Green, S. J., and Turberfield, A. J. (2005) Angew. Chem. Int. Ed. Engl. 44, 4358-4361.
- Ishibashi, S., Love, N. R., and Amaya, E. (2012) Meth. Mol. Biol. 917, 205-18.
- Ishibashi, S., Kroll, K. L., and Amaya, E. (2012) Meth. Mol. Biol. 917, 185-203.
- Marx, V. (2012) Nat. Methods 9, 1055-1059.
- Händel, E., and Cathomen, T. (2011) Curr. Gene Ther. 11, 28-37.
- Joung, J. K., and Sander, J. D. (2012) Nat. Rev. Mol. Cell Biol. 14, 49-55.
- Collin, J., and Lako, M. (2011) Stem Cells 29, 1021-1033.
- Carlson, D. F., et al. (2012) PNAS 109, 17382-17387.
- Ledford, H. (2011) Nature 471, 16.
- Gasiunas, G., et al. (2012) PNAS 109, E2579-2586.
- Jinek, M., et al. (2012) Science 337, 816-821.
