Epigenetics - Expanding on Genomic Foundations
Epigenetics is the study of heritable changes in the phenotype of a cell or organism that are not caused by its genotype. The molecular basis of an epigenetic profile arises from covalent modifications of protein and DNA components of chromatin. The epigenetic profile of a cell often dictates cell fate, as well as mammalian development, aging and disease. Epigenetic changes may persist for the remainder of a cell's life, but may also last for multiple generations in a lineage. Here, we provide an overview of the molecular basis for epigenetics and methods for studying DNA methylation.
Histone Modifications
In eukaryotes, chromatin is organized into nucleosome core particles that consist of approximately 147 bp of DNA and an octamer of histones (typically, two each of the core histones: H2A, H2B, H3 and H4) (1). The linker histone, H1, can further condense chromatin by binding to linker DNA between the nucleosome core particles. In mammals, chromatin can be generally classified as condensed, transcriptionally silent heterochromatin, or less-condensed, transcriptionally active euchromatin. Most genomic DNA is heterochromatin, which constitutes telomeres, pericentric regions and areas rich in repetitive sequences.
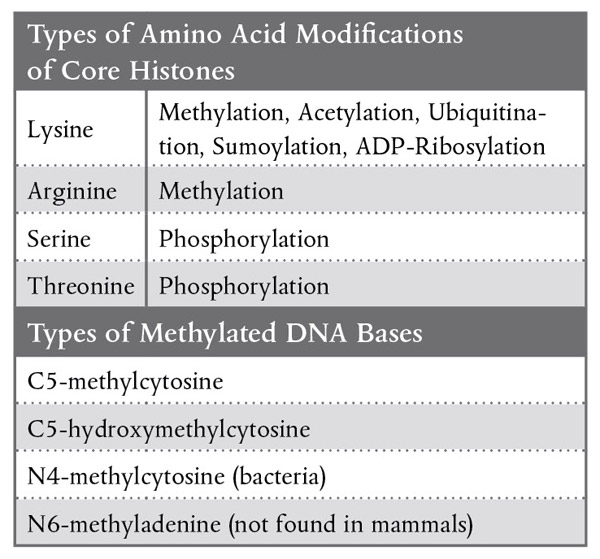
The core histones consist of a globular C-terminal domain and an unstructured N-terminal tail. Although a variety of modifications occur throughout the histone protein (Table 1), they occur primarily on the N-terminal tail. Some of these changes are enzymatically reversible. In general, the biological significance of all these modifications is not well understood, but the modifications are known to influence transcription, DNA repair, DNA replication and chromatin condensation. A "histone code" hypothesis is being tested by researchers to determine if combinations of histone modifications can be used to predict changes in gene expression (2-3). A comprehensive list of histone-modifying enzymes can be found in a review of mammalian epigenetic mechanisms by Kim et al (4). New England Biolabs offers a selection of unmodified, recombinant human histones (NEB#E5350) modifying enzymes (see product list).
Figure 1: The Epigenetic Code
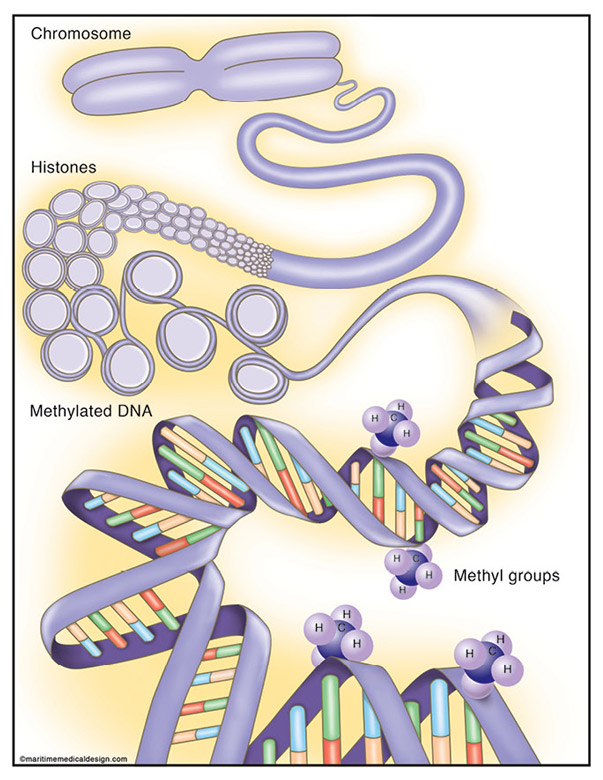
Table 1: Epigenetic Modifications
DNA Methylation
DNA can be modified by methylation of adenine and cytosine bases in a wide variety of prokaryotes and eukaryotes (Table 1). In prokaryotes, DNA methylation is involved in the determination of DNA-host specificity, virulence, DNA repair, chromosome replication and segregation, cell cycle regulation and gene expression. In higher eukaryotes, DNA methylation is involved in gene regulation, chromatin structure, differentiation, flowering, imprinting, mammalian X chromosome inactivation, carcinogenesis, complex diseases and aging.
DNA methylation in mammals primarily occurs on the fifth carbon of the cytosine base (5 methylcytosine, 5-mC) of CpG dinucleotides. Approximately 70% to 80% of CpG dinucleotides are methylated in somatic cells. However, 5-mC at CpA, CpT and CpC sequences have also been found in genomic DNA from mouse embryonic stem cells, and 5-mC at CpA sequences are thought to regulate enhancers in mouse brain. Of note, while DNA methylation in mammals primarily occurs at CpG dinucleotides, DNA methylation in plants may occur at CpG, CpHpG and CpHpH sequences, where H is adenine, cytosine, or thymine.
Recently, 5-hydroxymethylcytosine (5-hmC) was discovered in mouse embryonic stem cells, Purkinje neurons and granule neurons (5-6). The role of this modified base is not known, but it may be involved in demethylation, or it may influence chromatin structure and local transcriptional activity by either recruiting selective 5-hmC-binding proteins or excluding proteins that specifically bind 5-mC. The study of 5-hmC has been hampered, because it cannot be distinguished from 5-mC in many assays (e.g., enzymatic digestion, bisulfite treatment). Recently, an enzymatic method based on radiolabeled glucosylation of 5-hmC by a bacteriophage β-glucosyltransferase, was used to detect and quantitate 5-hmC in a sequence independent manner (7). Scientists at New England Biolabs have developed an enzymatic method that does not require the use of radioactivity, which is based on treatment of the DNA with T4 Phage β-glucosyltransferase, followed by cleavage with methylation-sensitive restriction enzymes (MspI and HpaII). This forms the basis of the EpiMark™ 5-hmC and 5-mC Analysis Kit. Additionally, while MeCP2 (a methylcytosine-binding protein, MBP) and antibodies against 5-mC specifically bind to 5-mC, similar reagents with specificities for 5-hmC have been developed.
Role of DNA Methylation in Mammals
DNA methylation in mammals influences a wide range of developmental and pathological processes. DNA methylation is required for normal embryonic development and survival of differentiated cells (8-10). Early in development, the paternal genome is actively demethylated, 4 and the maternal genome is subsequently demethylated, potentially through a passive mechanism. Methylation then increases in the blastocyst to generate the methylation patterns observed in adults. In addition, genomic imprinting, which is the specific expression of a paternal or maternal gene in placental mammals and is necessary for normal embryonic, neonatal and neurological growth, is mediated by DNA methylation and noncoding RNAs. These imprints are established during sperm and egg development by DNA methyltransferase Dnmt3A and are maintained by Dnmt1 isoforms. X inactivation, which is the process of mammalian dosage compensation of X-linked genes, is also mediated by DNA methylation and noncoding RNA.
Many cancers involve generalized, genomewide hypomethylation and local hypermethylation of CpG islands associated with promoters (reviewed in 4, 11). Demethylation of long interspersed nuclear elements (LINEs), which is a family of repetitive DNA sequences, occur early in some cancers, and the degree of LINE methylation is often correlated with the degree of malignancy. Cancer patients can vary in the frequency of methylation changes, and those with hypermethylation of multiple genes are proposed to have a CpG island methylator phenotype (CIMP), which could impact diagnosis, treatment and outcomes. In cancer, epigenetic changes are more frequent than genetic mutations and have resulted in cancer-specific biomarker discovery (e.g., Septin 9 for colorectal cancer). Although the significance of each epigenetic change is not clear, hundreds to thousands of genes can be epigenetically silenced by DNA methylation.
Increasing evidence suggests that epigenetic and genetic abnormalities also contribute to the development of other complex diseases such as type II diabetes, schizophrenia, autoimmune disease, hypertrophic cardiomyopathy, long QT syndrome and autism. Epigenetic mechanisms may help explain some features of complex diseases, including late onset, gender effects, parent-oforigin effects, phenotypic differences between monozygotic twins and fluctuation of symptoms.
Epigenetics in Clinical Applications
Epigenetic modifications and enzymes have the potential to be the basis of new therapeutics and diagnostic tests for diseases or syndromes with epigenetic components. So far, a histone deacetylase inhibitor and two DNA methyltransferase inhibitors (azanucleoside drugs) have been approved by the United States Food and Drug Administration to treat T cell cutaneous lymphoma and myelodysplastic syndrome, respectively. Additional drug candidates that inhibit histone deacetylases and DNA methyltransferases are in development (12-14), as are histone methyltransferase inhibitors and DNA methylation inhibitors that do not require incorporation into DNA like the azanucleoside drugs. The utility of combination therapies and development of more specific, targeted therapies remain areas of interest. In addition, because cancers are frequently associated with hypermethylated tumor suppressor genes and because tumor-derived DNA is present in various, easily accessible body fluids, methylated DNA could be a biomarker for detecting some cancers (15-18). Epigenetic therapies and biomarkers have also been studied and developed for systemic lupus erythematosis.
Methods to Study DNA Methylation
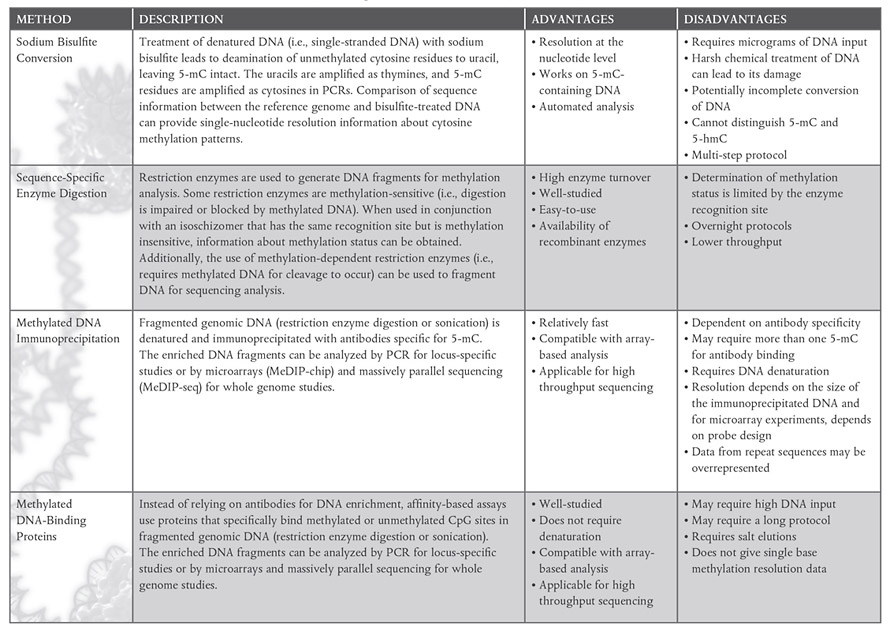
There are three main approaches for studying DNA methylation today. These are based on pretreating genomic DNA with either sodium bisulfite, restriction enzymes or a methylated DNA-binding affinity matrix (Table 2). Briefly, using sodium bisulfite to convert unmethylated cytosines to uracil, as opposed to 5-mC, which is refractory to bisulfite-mediated deamination, is the gold standard for assessing DNA methylation. This is partly because this technique can reveal the methylation status of every cytosine residue and is amenable to massively parallel sequencing methods. Differential enzymatic cleavage of DNA relies on methylation-sensitive or methylation-dependent restriction enzymes fragmenting genomic DNA for subsequent analysis. Reaction conditions used for restriction enzyme-based methods are easy-touse and not as harsh as those required for bisulfite methods; however, the resolution of the data is limited by the availability of enzyme recognition sequence. Finally, affinity-based methods use methylated DNA binding proteins or antibodies to enrich the experimental DNA sample for subsequent analysis.
A wide variety of analytical and enzymatic downstream methods can be used to characterize genomic DNA. Analytical methods, such as highperformance liquid chromatography (HPLC) and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS), have been used to quantify modified nucleobases in complex DNA. While HPLC is quantitative and reproducible, it requires large amounts of DNA and is often unsuitable for high throughput applications. MALDI-TOF MS is also quantitative, but is more amenable to high throughput applications. Other downstream methylation detection methods include end-point PCR, real-time PCR, primer extension, single-stranded conformational polymorphism assays, blotting, microarrays, and sequencing. Choosing which method(s) to use largely depends on the experimental sample size and the goals of the experiments (19). For more information about several of these methods, visit here.
Future Prospects and Challenges
The epigenetic code is hypothesized to be the combined effects of histone modifications and DNA methylation on gene expression (Table 1). While the genetic code in an individual is the same in every cell, the epigenetic code could be tissue- and cell-specific and may change over time because of aging, disease or environmental stimuli (e.g., nutrition, life style, toxin exposure) (reviewed in 20). Cross-talk between histone modifications, DNA methylation or RNAi pathways are being studied in such areas as cancer, X inactivation and imprinting.
Studying the timing and changes in epigenetic modifications during development and disease has many challenges. In addition, epigenome maps are still being assembled for most organisms. Advances in research methodologies must address issues such as the reduction in sample size requirements for histone modification studies and biomarker detection, development of better antibodies, development of more reagents and methods that can distinguish 5-mC and 5-hmC, improving highly parallel DNA analyses, and developing computational tools to organize and integrate diverse epigenomic data.
Table 2. Brief comparison of current methods to pretreat genomic DNA for methylation analysis.
References
- Kornberg, R.D. (1977) Annu. Rev. Biochem. 46, 931-954. PMID: 332067
- Strahl, B.D. and Allis, C.D. (2000) Nature, 403, 41-45. PMID: 10638745
- Jenuwein, T. and Allis, C.D. (2001) Science, 293, 1074-1080. PMID: 11498575
- Kim, J.K., Samaranayake, M. and Pradhan S. (2009) Cell. Mol. Life Sci. 66, 596-612. PMID: 18985277
- Kriaucionis, S. and Heintz, N. (2009) Science, 324, 929-930. PMID: 19372393
- Tahiliani, M., Koh, K.P., Shen, Y., et al. (2009) Science, 324, 930-935. PMID: 19372391
- Szwagierczak, A., Bultmann, S., Schmidt, C.S., Spada, F. and Leonhardt, H. (2010) Nuc Acids Res. doi:10.1093/nar/ gkq684. PMID: 20685817
- Li, E., Bestor, T.H. and Jaenisch, R. (1992) Cell, 69, 915-926. PMID: 1606615
- Okano, M., Bell, D.W., Haber, D.A. and Li, E. (1999) Cell, 99, 247-257. PMID: 10555141
- Jackson-Grusby, L., Beard, C., Possemato, R., et al. (2001) Nat. Genet. 27, 31-39. PMID: 11137995
- Jones, P. A. and Baylin, S. B. (2002) Nat. Rev. Genet. 3, 415-28. PMID: 12042769
- Bolden, J.E., Peart, M.J. and Johnstone, R.W. (2006) Nat. Rev. Drug Discov. 5, 769-784. PMID: 16955068
- Jones, P.A. and Taylor, S.M. (1980) Cell, 20, 85-93. PMID: 6156004
- Cheng, J.C., Yoo, C.B., Weisenberger, D.J., et al (2004) Cancer Cell, 6, 151-158. PMID: 15324698
- Qureshi, S.A., Bashir, M.U. and Yaqinuddin, A. (2010) Int. J. Surg. 8, 194-198. PMID: 20139036
- Kim, M.S., Lee, J. and Sidransky, D. (2010) Cancer Metastasis Rev. 29, 181-206. PMID: 20135198
- Despierre, E., Lambrechts, D., Neven, P., Amant, F., Lambrechts, S. and Vergote, I. (2010) Gynecol. Oncol. 117, 358-365. PMID: 20207398
- Martens, J.W., Margossian, A.L., Schmitt, M., Foekens, J. and Harbeck, N. (2009) Future Oncol. 5, 1245-1256. PMID: 19852739
- Laird, P.W. (2010) Nat. Rev. Genet. 11, 191-203. PMID: 20125086
- Tost, J. (2010) Mol. Biotechnol. 44, 71-81. PMID: 22821593

