Avoid Common Obstacles in Protein Expression
All too often, a protein of interest expresses poorly due to toxicity in the host cell, insolubility, or mRNA secondary structure preventing interactions with cellular machinery. Occasionally, the gene of interest is rich in codons that are inconsistent with the host strain's available supply of tRNAs. Uncontrolled basal expression can affect host cell growth and decrease protein yield, while overly robust induction can result in inclusion bodies. Exporting a protein to the E. coli periplasm or the inner membrane introduces more complications for targets that must be folded with disulfide bonds or incorporated into a membrane.
NEB has a long history in recombinant protein expression and has developed a breadth of knowledge that serves as a valuable resource for customers. Often a well-timed piece of advice is enough to fix experimental troubles. But for more difficult problems—or for simply streamlining the process and improving yield—NEB's portfolio of expression products offers a variety of solutions.
Choosing an E. coli Host Strain
E.coli strains are generally designed for cloning or for protein expression; although some strains are suitable for both purposes. The endA1 mutation is an important host feature for cloning and propagation of plasmid DNA since mutation of the endA gene abolishes Endonuclease I activity resulting in higher quality plasmid preparations. The T7 Express and NEB Express strains offer the option of direct cloning, followed by protein expression; these strains carry the endA1 mutation and are available as high efficiency competent cells (Table 1). The common expression strain BL21(DE3) is a poor choice for direct cloning, because its Endonuclease I activity may degrade plasmids after isolation, and its high basal T7 expression level may result in clone instability and/ or intolerance of toxic proteins. Another host feature to consider during cloning is the recA1 mutation, which abolishes homologous recombination. Undesired DNA recombination is more likely when the gene contains repeat sequences or if the plasmid clone contains sequences homologous to the host chromosome. Most cloning strains carry a recA mutation, while it is generally not necessary for protein expression strains unless the plasmid clone is known to be unstable.
Table 1. T7 Express and NEB Express Competent Cell Strains
| STRAIN | NEB # |
|---|---|
| NEB Express Competent E. Coli (High Efficiency) |
C2523H/I |
| NEB Express lq Competent E.Coli (High Efficiency) |
C3037H/I |
| T7 Express Competent E. Coli | C2566H/I |
| T7 Express lysY Compeent E. Coli (High Efficiency) | C3010H/I |
| T7 Express lysY/lq Competent E.Coli (High Efficiency) | C3013H/I |
With regard to expressed protein quality, researchers should seek expression strains that lack proteases, such as OmpT, which are likely to degrade target proteins during processing. Along with T7 Express and NEB Express, several commercially available strains lack the proteases OmpT and Lon. One caveat, however––Lon may serve as a "quality control" protease important to particular expression scenarios (1). For proteins that show signs of proteolysis, NEB recommends using an OmpT-deficient expression strain and adding protease inhibitor cocktail during processing.
Additionally, strains that lack F´ episomes are preferred as protein expression hosts because many cloning strains carry the ompP protease gene on an F´ episome (2). Four expression strains from NEB carry a miniF plasmid, but this single-copy vector lacks the ompP gene.
Many other attributes of good host strains, such as lacIq and lysY control of basal expression are addressed below in discussions of gene expression problems.
Figure 1: LysY is a T7 lysozyme variant (K128Y)
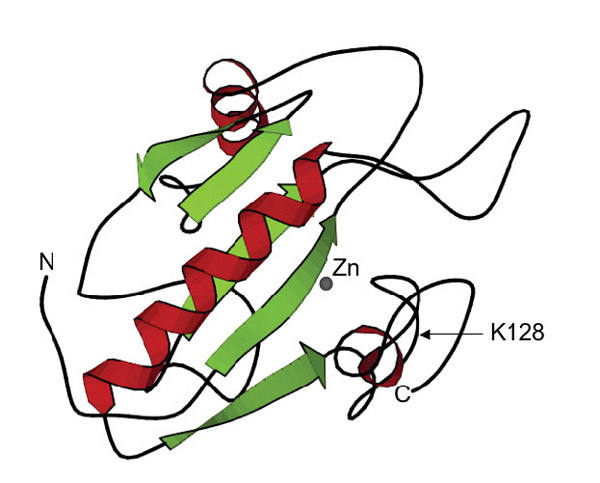
SWISS-Pdb viewer, PDB ID:1LBA)
Solving High Basal Expression in an IPTG-inducible System
High uninduced expression of a target protein can seriously hamper a host strain's viability or result in loss of plasmid from a significant share of the cell population.
Many commercial plasmids and host strains can provide regulated expression to reduce or eliminate this problem. For expression plasmids using a variation of the lac promoter, such as Plac, PlacUV5, Ptac and Ptrc, the first step is to be certain that the expression system supplies additional LacI repressor. Most systems include the lacI gene on an expression vector, while many host strains feature enhanced LacI production (3).
Additional plasmid encoded lacI genes are often not enough to control basal expression; so many systems instead supply the lacIq gene, whose mutated promoter increases LacI repressor expression ten-fold (4). NEB scientists recommend using a host expression strain harboring the lacIq gene (e.g., NEB Express Iq). Compared to strains lacking lacIq, strains carrying this gene are more easily transformed with lac-promoter or T5-lac promoter plasmids carrying genes that encode toxic proteins (3).
Reducing Basal Expression in the T7 System
The most common protein-expression strain, BL21(DE3), expresses T7 RNA polymerase at a high basal level. Target proteins in this strain and many of its derivatives are often expressed before inducer is added.
Control of T7 expression is best provided by hosts that co-express T7 lysozyme, which naturally inhibits T7 RNA Polymerase through a 1:1 protein interaction (5). T7 lysozyme is expressed in plasmids pLysS and pLysE, as well as in lysY host strains. Researchers at NEB recommend switching to a lysY or pLysS strain as a first resort if plasmid transformation fails, or if the protein of interest might be toxic to the host.
Plasmids carrying lysS or lysE produce T7 lysozyme with amidase activity. A freeze-thaw cycle can lyse strains carrying pLysS, so it's important to consider downstream processing when planning T7-system expression. LysY host strains produce a variant T7 lysozyme that lacks amidase activity.
It is important for investigators to take care when using pLysE hosts—the excess amidase activity of pLysE can damage E. coli cell walls, which may cause a growth defect (6). NEB researchers have observed culture lysis in experiments with pLysE strains, in which the protein of interest is targeted to the cell envelope.
In DE3 strains, adding 1 percent glucose to the medium can decrease basal expression from the lacUV5 promoter by lowering the cAMP levels that stimulate it. Switching from glucose to carbon source, such as glycerol, in final growth cycles can also help maximize IPTGinduced expression (6).
Low-Basal Expression Alternatives
NEB's T7 Express strains use a wild-type lac promoter to express the T7 RNA polymerase from within the lac operon, which results in lower basal production compared to DE3 inistrains. T7 Express strains also optionally express lacIq , lysY, or a combination of these control elements.
Tunable Expression for Toxic Proteins
In addition to tight promoter control, expressing toxic proteins often requires tunable expression. Keeping expression at a desired moderate level can maximize yields by maintaining the concentration of a toxic target protein just below a host strain's tolerance. Alternatively, tuning expression allows researchers to prevent well-expressed target proteins from creating inclusion bodies.
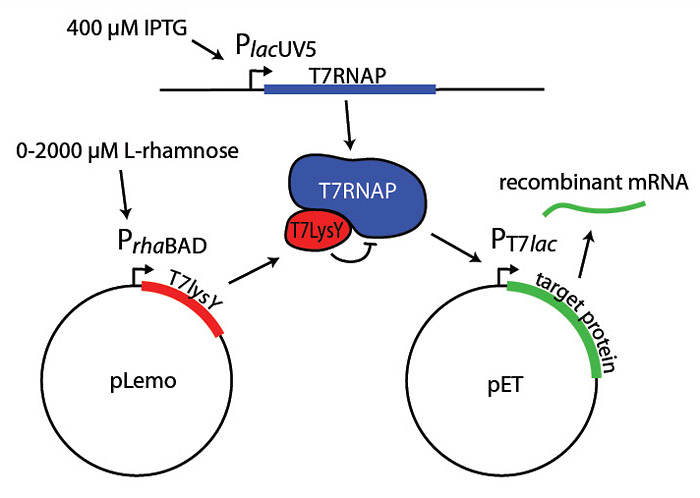
The PrhaBAD promoter is a key part of many expression systems. For example, the Lemo21(DE3) strain expresses lysY control protein under the PrhaBAD promoter (Fig 2). Finding the right expression level involves running parallel expression trials using L-rhamnose concentrations from 0 µM to 2,000 µM. While most promoters are either "on" or "off," protein production per cell in the Lemo21(DE3) strain is inversely proportional to L-rhamnose concentration.
To express a highly toxic protein, it may be necessary to employ a cell-free expression system, such as the PURExpress In Vitro Protein Synthesis Kit, which uses only recombinant components, and is free of contaminating nucleases, proteases, and proteinmodifying enzymes.
Figure 2. Tuning protein expression in Lemo21(De3)
Raising Low-Solubility Protein Yields
Proteins that are insoluble—or nearly insoluble— require approaches beyond tuning expression. Inducing protein expression at a lower temperature, between 15–20°C, can often raise yields of properly folded protein.
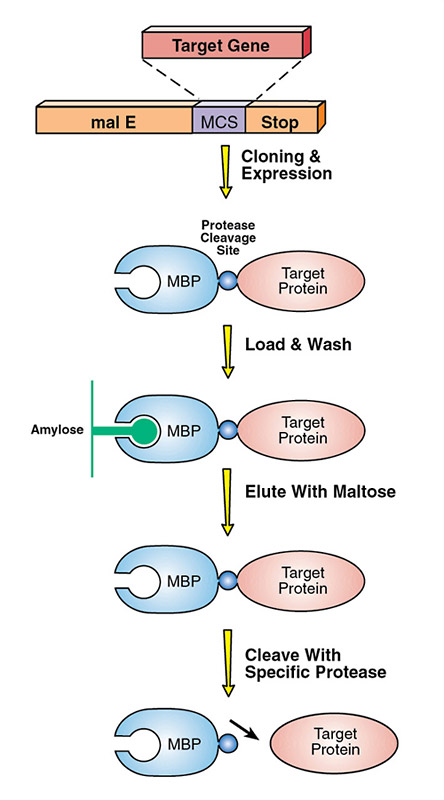
Another approach to expressing low-solubility proteins is to fuse them to a "solubility tag" using vectors such as the pMAL Protein Fusion and Purification System. The pMAL vectors encode maltose binding protein, a fusion tag that aids in expression and solubility, and allows for simple purification using an amylose column (Fig 3). Removing the MBP fusion requires protease cleavage and additional purification. However many MBP fusion proteins can be readily studied, since they often retain activity in this form.
Some researchers achieve good results by coexpressing low-solubility proteins with chaperonins, such as GroEL, DnaK, and ClpB (7,8). While chaperonin overexpression may improve target protein solubility, some target protein may remain complexed with chaperones. Methods including native PAGE analysis and size exclusion chromatography can reveal oligomeric complexes in expressed protein samples.
Figure 3. Schematic of the pMal System
Changing Sequence to Improve Expression
During recombinant gene expression experiments, intra-RNA interactions can sometimes prevent optimal translation. Troublesome secondary structure can be a problem in the 5´ untranslated region, the ribosomal binding site and the affinity tag coding sequence. Even the frequency of particular codons in the gene of interest can cause expression problems.
In genes with troublesome secondary structure, it is often possible to improve expression by altering ribosomal binding sites and removing inhibitory secondary structure. Altering ribosomal binding sites for better expression usually means changing their sequences to more closely match the ideal E. coli sequence, AGGAGGT. Changing an affinity tag's position, and adding more adenines to the next codon after the initiation codon (9), may also improve expression in some cases.
Finally, translation can stall in genes whose translation calls for tRNAs that are in low abundance in the host species (10). In this case, consider co-expressing rare tRNAs in the host organism (11) or completely redesign the gene using preferred bacterial codons. The decreasing cost of gene synthesis makes this second option increasingly attractive. However, redesigned genes can become so well-expressed that solubility and inclusion bodies begin to become problems, and it may be necessary to adopt a tunable expression system.
Making the Right Disulfide Bonds
When disulfide bonds are essential for target protein folding or stability, investigators often direct the protein to E. coli's oxidative periplasm, where Dsb enzymes can establish the correct bond configuration. Several commercially available vectors include an N-terminal signal sequence for exporting proteins to the periplasm. An example would be the pMALp5 vectors, whose wild-type MBP gene contains an N-terminal periplasmic localization signal.
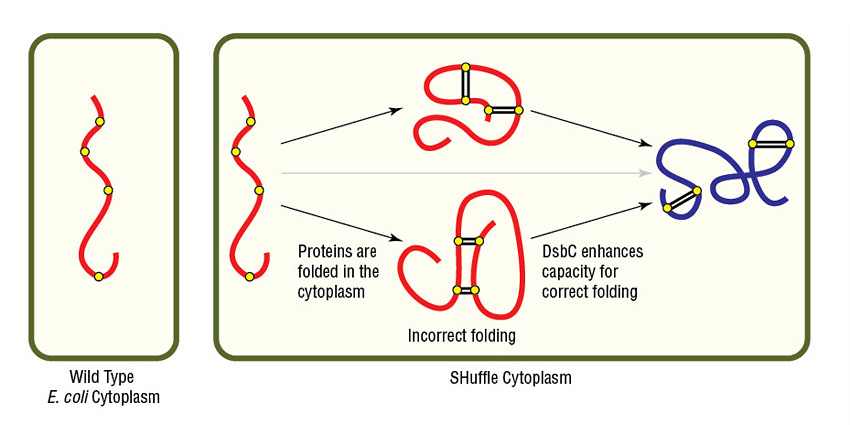
Alternatively, NEB's SHuffle strains are excellent options for expressing proteins having complex disulfide bonds. SHuffle strains carry mutations that alter cellular reduction conditions, allowing proper disulfide bond formation in a now-partially oxidizing cytoplasm (Fig 4). SHuffle strains also express disulfide bond isomerase (DsbC) in the cytoplasm, rather than only in the periplasm.
It is also possible to modify cell-free systems to produce proteins with disulfide bonds. In one approach, eliminating DTT from the reaction mixture before translation can properly alter oxidation conditions for bond formation (12). A second approach adds iodoacetamide, a glutathione redox buffer, and a disulfidebond isomerase (13). For researchers using the PURExpress system, the PURExpress Disulfide Bond Enhancer— a proprietary blend of proteins and buffer components—will assist correct folding of proteins with multiple disulfide bonds.
Figure 4. Expression of protein with multiple disulfide bonds using SHuffle Competent E. coli
Improving Membrane-Protein Yields
Membrane proteins are especially difficult to produce in quantity, and targeting them to the E. coli inner membrane is often the best expression strategy. However, in unregulated expression systems it is possible for newly synthesized protein to overwhelm the SecYEG translocation machinery. These situations often require a tunable expression system, such as Lemo21(DE3) Competent E. coli (14).
As another solution to translocation bottlenecks, NEB researchers have weakened the ribosomal binding site by altering its sequence, which lowered basal expression and enabled a higher yield of a difficult membrane protein (3).
Difficult membrane proteins may also call for a low- to medium-copy plasmid conferring kanamycin or chloramphenicol resistance— rather than ampicillin resistance—to reduce the likelihood of plasmid loss. NEB researchers recommend testing for expression plasmid maintenance at the point of induction by plating cells with and without antibiotic. After each expression experiment, verify that a significant portion of the target membrane protein is integrated into the membrane. If not, express the protein at a lower temperature, perhaps 20–25°C, and with early induction at OD600 of 0.35–0.45.
As an alternative to host-cell expression, cellfree translation systems are also viable options for expressing troublesome membrane proteins. Ion-channel proteins, transporters, receptors, and other integral membrane proteins can affect viability by disrupting membranes, or simply by aggregating in the cytoplasm. Either case leads to low yields.
Cell-free expression of membrane proteins often requires additional detergents, synthetic lipids or bilayers similar to those in a target protein's source organism. When aggregation is a problem, cell-free systems allow researchers to investigate adding detergents or lipids to prevent precipitation.
Future of Protein Expression Techniques
Some proteins present truly intractable expression problems in heterologous hosts. But protein expression techniques are currently experiencing rapid improvement, with new developments in tunable expression, solubility technology, protein targeting, and cell-free systems, greatly improving yields and purity over the expression systems of only a few years ago. NEB aims to continue contributing to the field with innovative Protein Expression products and helpful advice.
References
- Link, A. J., et al. (2008) Protein Sci. 17, 1857–1863.
- Hwang, B. Y., et al. (2007) J Bacteriol. 189, 522–530.
- Samuelson, J. C. (2011) Recent Developments in Difficult Protein Expression: A Guide to E. coli Strains, Promoters, and Relevant Host Mutations. Methods Mol. Biol. 705:195–209.
- Calos, M. P. (1978) Nature, 274, 762–765.
- Zhang, X. and Studier, W. F. (1997) J. Mol. Biol. 269, 10–27.
- Pan, S. H. and Malcolm, B. A. (2000) Biotechniques, 29, 1234–1237.
- Amrein, K.E., et al. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 1048–1052.
- Nishihara, K., et al. (1998) Appl. Environ. Microbiol. 64, 1694–1699.
- Stenstrom, C.M., et al. (2001) Gene, 263, 273–284.
- McNulty, D.E., et al. (2003) Protein Expr. Purif. 27, 365–374.
- Dieci, G., et al. (2000) Protein Expr. Purif. 18, 346–354.
- Kawasaki, T. et al. (2003) Eur. J. Biochem. 270, 4780– 4786.
- Yin, G. and Swartz, J.R. (2004) Biotechnol. Bioeng. 86, 188–195.
- Wagner, S., et al. (2008) Proc. Natl. Acad. Sci. USA 105, 14371–14376.
From NEB expressions Summer 2011
Article by Chris Womack, Science Writer, Austin, TX

