sgRNA Synthesis Using the HiScribe®; Quick T7 High Yield RNA Synthesis Kit (NEB #E2050)
Single guide RNAs (sgRNAs) are incorporated into Cas9 ribonucleoprotein complexes (RNPs) and function to direct sequence-specific DNA binding. The resulting programmed Cas9 RNPs can be used to cleave (or bind, or nick) dsDNA. More information on applications of Cas9 RNPs can be found here.
The HiScribe T7 Quick High Yield RNA Synthesis Kit is designed for quick setup and production of large amounts of RNA in vitro. The reaction can be set up conveniently by combining the NTP Buffer Mix, T7 RNA Polymerase Mix and a suitable DNA template.
DNA Template PreparationsgRNA transcription templates contain a T7 RNA Polymerase promoter followed by an ~20 nt target-specific region and an ~80bp constant region (Figure 1). Target specific regions can be predicted using online tools such as CHOPCHOP or Benchling. The constant region of the sgRNA is required for interaction with Cas9 protein.
Schematic representation of sgRNA transcription templates

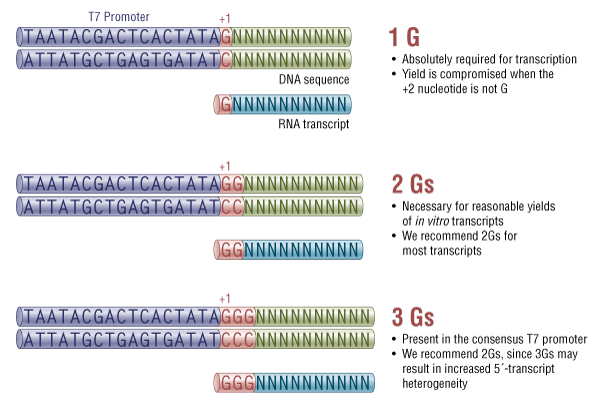
Note that because of the sequence requirements of the T7 RNA Polymerase promoter, we recommend that the first 2 nucleotides of the sgRNA transcript are GG (Figure 2). This region of the sgRNA plays a relatively minor role in targeting [1-4]. If an sgRNA target-specific regions begins with one or more Gs, these can take the place the recommended GG in the suggested template. We recommend that the final template is designed so that transcription initiates with at least 2, and ideally only 2 Gs. An example sgRNA template can be found in FASTA format below.
T7 promoter sequence requirements
Linearized plasmid DNA, PCR products or synthetic DNA oligonucleotides can be used as templates for in vitro transcription with the HiScribe T7 Quick High Yield RNA Synthesis Kit, provided they contain the T7 promoter upstream of the sequence to be transcribed.
Synthetic DNA Oligonucleotides are a rapid and convenient way to generate sgRNA transcription templates. We recommend that the DNA oligonucleotide templates be completely double stranded. Partially double-stranded oligonucleotide templates with a double-stranded T7 promoter sequence may be used in the T7 Quick High Yield RNA Synthesis Kit, but yields may be variable and in general are lower than when fully double stranded templates are used.
gBlocks® Gene Fragments from Integrated DNA Technologies are sequence verified double-stranded synthetic DNA fragments. These custom gene fragments, or duplexed Ultramer® Oligonucleotides are a good source for synthetic transcription templates. Note that it is important to order both strands when ordering Ultramers.
We recommend adding a few nucleotides 5´- to the T7 promoter when oligonucleotides templates are used to minimize the effect of n-1 oligonucleotide synthesis products, which can result in impaired promoter function.
Use ~ 1 picomole (75 nanograms of fully double-stranded synthetic oligonucleotide) for each 30 µL RNA synthesis reaction.
Plasmid TemplatesPlasmid templates for sgRNA synthesis can be obtained from a number of sources, including Addgene (search for gRNA vectors with T7 promoters)]. Empty gRNA vectors can be modified to encode target-specific sgRNAs using the Q5® Site-Directed Mutagenesis Kit, restriction enzyme cloning, or the NEBuilder® HiFi Assembly Master Mix (see application note).
Quality of the template DNA affects transcription efficiency, as well as the integrity of the RNA synthesized. It is therefore critical to begin the protocol with highly purified DNA. Any plasmid purification method may be used, as long as the product is predominately supercoiled and free of contaminating RNase, protein, RNA and salts.
To produce an RNA transcript of defined length, plasmid DNA must be completely linearized with a restriction enzyme downstream of the insert to be transcribed. NEB has a large selection of restriction enzymes for this purpose; we recommend selecting restriction enzymes that generate blunt ends or 5´-overhangs.
After linearization, we recommend purifying the template DNA by phenol:chloroform extraction:
- Extract DNA with an equal volume of 1:1 phenol:chloroform mixture.
- Extract twice with an equal volume of chloroform to remove residual phenol.
- Precipitate the DNA by adding 1/10th volume of 3 M sodium acetate, pH 5.2, and two volumes of ethanol. Incubate at –20°C for at least 30 minutes.
- Pellet the DNA in a microcentrifuge for 15 minutes at top speed. Carefully remove the supernatant.
- Rinse the pellet by adding 500 μl of 70% ethanol and centrifuging for 15 minutes at top speed. Carefully remove the supernatant.
- Air dry the pellet and resuspend it in nuclease-free water at a concentration of 0.5–1 μg/μl.
We recommend using about 1.5 micrograms of plasmid template (~1 picomole of a 2.5 kb plasmid) for each 30 µL RNA synthesis reaction.
PCR TemplatesTranscription templates for sgRNA synthesis can be PCR amplified from plasmid or synthetic oligonucleotide templates and appropriate PCR primers. We recommend the use of Q5 High Fidelity Hot Start 2X Master Mix for generation of PCR templates.
PCR products should be run on an agarose gel to estimate concentration and to confirm amplicon size prior to its use as a template in the T7 Quick RNA transcription reaction.
PCR mixture may be used directly if diluted at least 10X in the transcription reaction. However, better yields will be obtained with purified PCR products. PCR products can be purified according to the protocol for plasmid restriction digests above, or by using commercially available spin columns.
Generally, 1 picomole (about 75 ng of a 120 bp PCR product) can be used in a 30 μl in vitro transcription reaction.
sgRNA SynthesisWe strongly recommend wearing gloves and using nuclease-free tubes and reagents to avoid RNase contamination. Reactions are typically 30 μl but can be scaled up as needed. Reactions should be assembled in nuclease-free microfuge tubes or PCR strip tubes.
- Thaw the necessary kit components, mix and pulse-spin in microfuge to collect solutions to the bottoms of tubes. Keep on ice.
- Assemble the reaction at room temperature in the following order:
| Nuclease-free water | X µl |
| NTP Buffer Mix | 10 µl (6.7 mM each NTP final) |
| Template DNA | X µl (1.5 µg (1 picomole of 2.5 kb plasmid))
or X µl (75 ng (1 picomole of 120 bp PCR product)) or X µl (75 ng (1 picomole of 120 bp synthetic dsDNA oligonucleotide)) |
| T7 RNA Polymerase Mix | 2 µl |
| Total reaction volume | 30 µl |
- Mix thoroughly and pulse-spin in a microfuge.
- Incubate at 37°C for 4 hours or longer for maximum yield. It is safe to incubate the reaction for 16 hours (overnight), however, sgRNA amounts sufficient for many experiments may be synthesized in less than 4 hours. We recommend incubation in a dry air incubator or a thermocycler to prevent evaporation of the sample.
Note that sgRNA synthesis reactions use 10 μl more water than standard HiScribe T7 Quick High Yield RNA Synthesis Kit reactions. The volume of NTP Buffer Mix and T7 RNA Polymerase Mix, however, remains the same.
Optional: DNase treatment to remove DNA template. Typical RNA synthesis reactions using the HiScribe T7 Quick High Yield RNA Synthesis Kit can generate large amounts of RNA, at concentrations up to 10 mg/ml. As a result, the reaction mixture can be quite viscous. It is easier to perform DNase treatment after diluting the reaction. To remove template DNA, add 20 μl nuclease-free water to each 30 μl reaction , followed by 2 μl of DNase I (RNase-free), mix and incubate for 15 minutes at 37°C.
- Proceed with purification of synthesized sgRNA or analysis of transcription products by gel electrophoresis.
Spin Column Purification
Spin columns will remove unincorporated nucleotides, proteins and salts. Please ensure that purification columns are compatible with size of sgRNAs (~80-100 nt) and follow the manufacturer’s instructions. We recommend the Monarch RNA Cleanup Kit (50 µg) (NEB #T2040).
Evaluation of sgRNA transcription productsQuantification by UV Light Absorbance
sgRNA concentration can be determined by measuring the ultraviolet light absorbance at 260 nm, however, any unincorporated nucleotides and template DNA in the mixture will affect the reading. Free nucleotides from the transcription reaction must be removed before the sgRNA concentration can be quantified. RNA solution can be read directly on a Nanodrop™ Spectrophotometer. The Nanodrop Spectrophotometer can directly read RNA concentrations from 10 ng/μl to 3000 ng/μl. For single-stranded RNA, 1 A260 is equivalent to an RNA concentration of 40 μg/ml. The RNA concentration can be calculated as follows:
A260 x dilution factor x 40 = __ μg/ml RNA
Alternatively, sgRNA concentration can be determined using a fluorometer, such as the Qubit® Fluorometer.
Analysis of Transcription Products by Gel Electrophoresis
To evaluate transcript length, integrity and quantity, an aliquot of the transcription reaction should be run on a denaturing polyacrylamide gel. The gels should be run under denaturing conditions to minimize formation of secondary structures by the transcript.
- We recommend using commercially available premade gels, such as Novex® 6% TBE-Urea Gels. Use standard TBE gel running buffer.
10X TBE buffer: 0.9 M T ris Base, 0.9 M Boric Acid, 20 mM EDTA. - Gel electrophoresis
a. Mix 0.2–1 μg RNA sample with 5-10 μl of RNA Loading Dye (2X, NEB #B0363).
b. Denature the RNA sample and an aliquot of RNA marker by heating at 65–70°C for 5–10 minutes.
c. Pulse-spin prior to loading onto gel.
d. Visualize RNA by staining the gel with SYBR® Gold or ethidium bromide.
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31: 827–832. doi:10.1038/nbt.2647
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339: 819–823. doi:10.1126/science.1231143
- Jiang W, Cox D, Zhang F, Bikard D, Marraffini LA. rNA-guided editing of bacterial genomes using crisPr-cas systems. Nat Biotechnol. Nature Publishing Group; 2013;31: 233–239. doi:10.1038/nbt.2508
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337: 816–821. doi:10.1126/science.1225829
>sgRNA-template
aagcTAATACGACTCACTATAGGnnnnnnnnnnnnnnnnnnnGTTTTAGAGCTAGAAATAGCAAGTTAAAATAA
GGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTT
Q5® and NEBUILDER® are registered trademarks of New England Biolabs, Inc.
HISCRIBE™ is a trademark of New England Biolabs, Inc.
gBLOCKS® and ULTRAMER® are registered trademarks of Integrated DNA Technologies, Inc.
RNA CLEAN AND CONCENTRATOR™ is a trademark of Zymo Research
NANODROP™ is a trademark of Thermo Fisher Scientific
QUBIT® and SYBR® are registered trademarks of Thermo Fisher Scientific

