Size Selection using AMPure XP Beads
Note: There are two different protocols for using AMPure XP beads. One is if you are first cleaning up your sample with a PCR column purification kit. The other option is to proceed directly after PCR.
Protocol
- Purify the PCR amplified cDNA construct (100 μl) using a QIAQuick PCR Purification Kit.
- Elute amplified DNA in 27.5 μl Nuclease-free Water.
- Load 1 μl of the purified PCR reaction on the Bioanalyzer using a DNA 1000 chip according to the manufacturer's instructions (Figure 1).
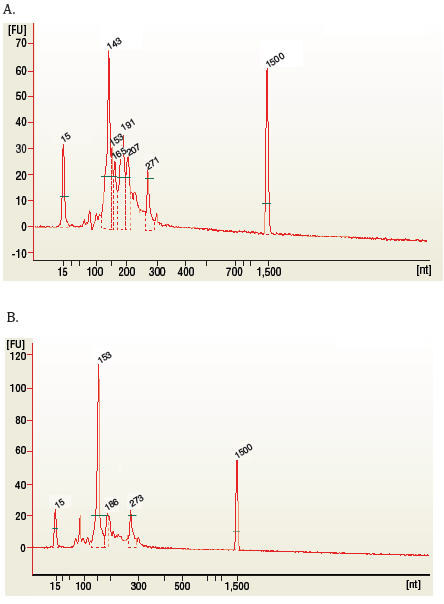
Figure 1: Typical results from (A) human brain and (B) rat testis total RNA libraries before size selection.
The 143 and 153 bp bands correspond to miRNAs and piRNAs, respectively. The bands on the Bionalyzer electropherograms resolve in sizes ~6-8 nucleotides larger than sizes observed on PAGE gels and can shift from sample to sample due to an incorrect identification of the marker by the bioanalyzer software. miRNA peak should be ~ 143-146 bp.
- To the purified PCR reaction (25 μl), add 32.5 μl (1.3X) of resuspended AMPure XP beads and mix well on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Place the tube on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully transfer the supernatant (57.5 μl) to a new tube (Caution: do not discard the supernatant). Discard beads that contain the large DNA fragments.
- Add 92.5 μl (3.7X) of resuspended AMPure XP beads to the supernatant (57.5 μl), mix well and incubate for 5 minutes at room temperature.
- Place the tube on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets (Caution: do not discard beads).
- Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 9 once.
- Briefly spin the tube, and put the tube back in the magnetic stand.
- Completely remove the residual ethanol, and air dry beads for 10 minutes while the tube is on the magnetic stand with lid open.
- Elute the DNA target from the beads with 15 μl nuclease-free water. Mix well on a vortex mixer or by pipetting up and down, and put the tube in the magnetic stand until the solution is clear.
- Transfer the supernatant to a clean PCR tube.
- Run 1 μl on the Bioanalyzer High Sensitivity chip. Check peak distribution and molarity of the small RNA library.
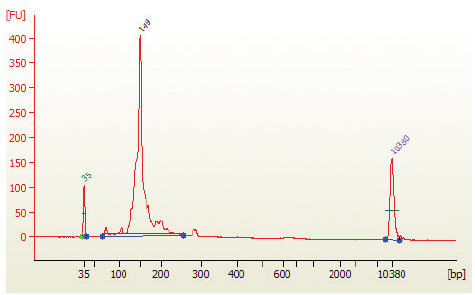
Figure 2: Electropherogram trace of the bead size selected purified library from human brain total RNA.
Size Select the small RNA library using AMPure XP beads (no column purification)
Note: Check the volume of the sample after PCR. Adjust the volume with nuclease free water if necessary to bring the volume up to 100 μl.- Transfer 100 μl sample to a 1.5 ml tube. Add 130 μl (1.3X) of resuspended AMPure XP beads and mix well on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Place the tube on an appropriate magnetic stand to separate the beads from the supernatant. After the solution is clear (about 5 minutes), carefully transfer the supernatant (230 μl) to a new tube (Caution: do not discard the supernatant). Discard the beads that contain the large DNA fragments.
- Add 370 μl (3.7X) of resuspended AMPure XP beads to the supernatant (230 μl), mix well and incubate for 5 minutes at room temperature.
- Place the tube on an appropriate magnetic stand to separate the beads from the supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets (Caution: do not discard beads).
- Add 1ml of freshly prepared 80% ethanol to the tube while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 6 once.
- Briefly spin the tube, and put the tube back in the magnetic stand.
- Completely remove the residual ethanol, and air dry beads for 10 minutes while the tube is on the magnetic stand with lid open.
- Elute the DNA target from the beads with 15 μl of nuclease-free water. Mix well on a vortex mixer or by pipetting up and down, and put the tube in the magnetic stand until the solution is clear.
- Transfer the supernatant to a clean PCR tube.
- Run 1 μl on the Bioanalyzer High Sensitivity chip. Check peak distribution and molarity of the small RNA library.
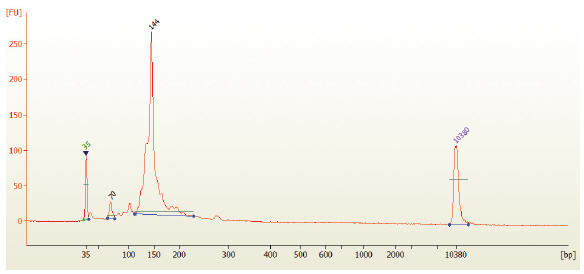
Figure 3: Electropherogram trace of the bead size selected library from human brain total RNA.
- Mix the purified PCR product (25 μl) with 5 μl of Gel Loading Dye, Blue (6X).
- Load 5 μl of Quick-Load pBR322 DNA-MspI Digest in one well on the 6% PAGE 10-well gel.
- Load two wells with 15 μl each of mixed amplified cDNA construct and loading dye on the 6% PAGE 10-well gel.
- Run the gel for 1 hour at 120 V or until the blue dye reaches the bottom of the gel. Do not let the blue dye exit the gel.
- Remove the gel from the apparatus and stain the gel with SYBR Gold nucleic acid gel stain in a clean container for 2–3 minutes and view the gel on a UV transiluminator (Figure 4).
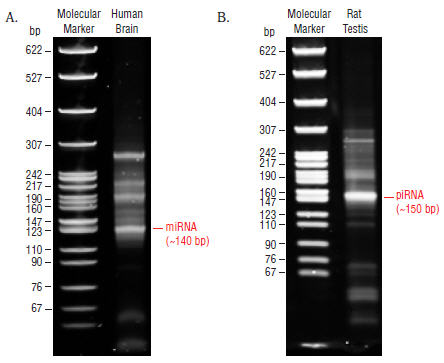
Figure 4: Shows typical results from Human Brain (A) and Rat Testis (B) Total RNA libraries.
The 140 and 150 bp bands correspond to miRNAs (21 nt) and piRNAs (30 nt), respectively. - The 140 and 150 nucleotide bands correspond to adapter-ligated constructs derived from the 21 and 30 nucleotide RNA fragments, respectively. For miRNAs, isolate the bands corresponding to ~140 bp. For piRNAs, isolate the band corresponding to ~150 bp.
- Place the two gel slices from the same sample in one 1.5 ml tube and crush the gel slices with the RNase-free Disposable Pellet Pestles and then soak in 250 μl DNA Gel Elution buffer (1X).
- Rotate end-to-end for at least 2 hours at room temperature.
- Transfer the eluate and the gel debris to the top of a gel filtration column.
- Centrifuge the filter for 2 min at > 13,200 rpm.
- Recover eluate and add 1 μl Linear Acrylamide, 25 μl 3M sodium acetate, pH 5.5 and 750 μl of 100% ethanol.
- Vortex well.
- Precipitate in a dry ice/methanol bath or at -80°C for at least 30 minutes.
- Spin in a microcentrifuge @ > 14,000 x g for 30 minutes at 4°C.
- Remove the supernatant taking care not to disturb the pellet.
- Wash the pellet with 80% ethanol by vortexing vigorously.
- Spin in a microcentrifuge (> 14,000 x g) for 30 minutes at 4°C.
- Air dry pellet for up to 10 minutes at room temperature to remove residual
ethanol. - Resuspend pellet in 12 μl TE Buffer.

