
Bisulfite sequencing, but at what cost?
Posted on Thursday, February 17, 2022
By
Topic: Tips for the lab
Bisulfite conversion intrinsically damages DNA resulting in patchy genome coverage that can be frustrating to deal with. Your methylation data may not what you wanted with some genome regions underrepresented, or even having incomplete conversion. Bisulfite conversion degrades DNA samples causing biased coverage and higher sequencing costs.
Let’s examine why so many core sequencing labs decline to perform Bisulfite-seq — even though intensive computational strategies exist to compensate for the inadequacies of bisulfite converted libraries. Then we can look at how Enzymatic Methyl-seq (EM-seq) bypasses the risks of bisulfite conversion to produce more accurate, sensitive, and biologically meaningful results with a variety of DNA methylation analysis techniques, as supported in recent scientific works.
Bisulfite conversion smells bad and yields methylome blind spots
In theory, using sodium bisulfite is a great approach to detect DNA methylation. Yet, how closely do its results reflect methylomes in vivo? The short answer is that biased coverage is a big problem.
In principle, it works by converting unmethylated cytosine to uracil, but not converting cytosines that are methylated (5mC) or hydroxymethylated (5hmC). Sodium bisulfite sulfinates unmethylated cytosines at a low pH, which causes a spontaneous hydrolysis reaction that removes an amino group from cytosine converting it into uracil, releasing ammonia in the process. (It’s clever chemistry but the ammonia part stinks!) PCR amplification will then convert uracil to thymine, so non-methylated cytosines are sequenced as thymines, and 5mCs or 5hmCs are sequenced as cytosines. Based on the C to T conversion of unmethylated cytosines we can precisely identify modified cytosines at single base resolution.
In practice, there are well known risks to using bisulfite treatment for methylome sequencing. Sample loss is intrinsic to the protocol. How that sample loss occurs adds barriers to scientific success. Bisulfite conversion requires extreme temperatures and pH, which causes depyrimidination of DNA, resulting in degradation. When sequencing adaptors are ligated prior to bisulfite conversion, the damage is initially observed with lower-than-expected bisulfite library yields. Unmethylated cytosines are damaged disproportionately compared to 5mC or 5hmC, resulting in sequencing blind spots and an unbalanced nucleotide composition. This is particularly evident in high GC content regions of genomes resulting in biased genome coverage. Addition of sequencing adaptors after bisulfite conversion to some extent recovers library yields - but biases do remain. The real risks with bisulfite conversion chemistry are low yields, uneven genome coverage and a biased GC distribution.
EM-seq bypasses bisulfite DNA damage without changing downstream analysis
Researchers can produce more biologically meaningful methylome data using enzymatic methyl conversion which was designed by NEB scientists to bypass bisulfite conversion risks and lessen the number of sequencing reads required, without disrupting downstream analysis sequencing pipelines.
In EM-seqTM, two sequential enzymatic reactions differentiate cytosine from its methylated and hydroxymethylated forms. The first reaction protects 5mC and 5hmC but not cytosines from deamination by APOBEC in the second reaction. By deaminating only cytosines - the modified forms can be identified by sequencing C as T and 5mC or 5hmC as C. Critically, notice how your DNA samples are not exposed to the extremes of temperature and the pH necessary for bisulfite conversion. Minimizing DNA damage upfront significantly promotes methylome data accuracy, especially with difficult and low input samples. The EM-seq method combines that enzymatic conversion technique with the NEBNext® Ultra IITM library preparation workflow reagents, with a range of inputs from 10 – 200 ng. The resulting high-quality EM-seq libraries enable superior detection of 5mC and 5hmC at single base resolution.
Very conveniently, EM-seq sequencing data can be processed using data analysis pipelines already established for bisulfite libraries. It’s by design that both bisulfite and EM-seq libraries sequence cytosines as thymines, and 5mC or 5hmC as cytosines. Reducing sample damage and the number of sequencing reads required wouldn’t be as helpful if EM-seq confounded downstream analysis, would it?
EM-seq retains sample complexity and reduces sequencing costs
Development of next generation sequencing (NGS) libraries focusses on accurately maintaining DNA sample complexity. Longer reads are desirable because they enable greater depth of sequencing and can cut costs. EM-seq beats bisulfite-seq in the metrics that matter most to achieve these goals.
To begin with, library yields for EM-seq are higher than for Whole Genome Bisulfite Sequencing (WGBS). This metric translates consistently to lower duplicates across input ranges. EM-seq libraries are typically longer than WGBS libraries. Standard EM-seq libraries are approximately 370-420 bp but insert sizes up to ~550 bp can also be achieved. These insert sizes enable longer sequencing reads which improve accuracy while reducing sequencing costs.
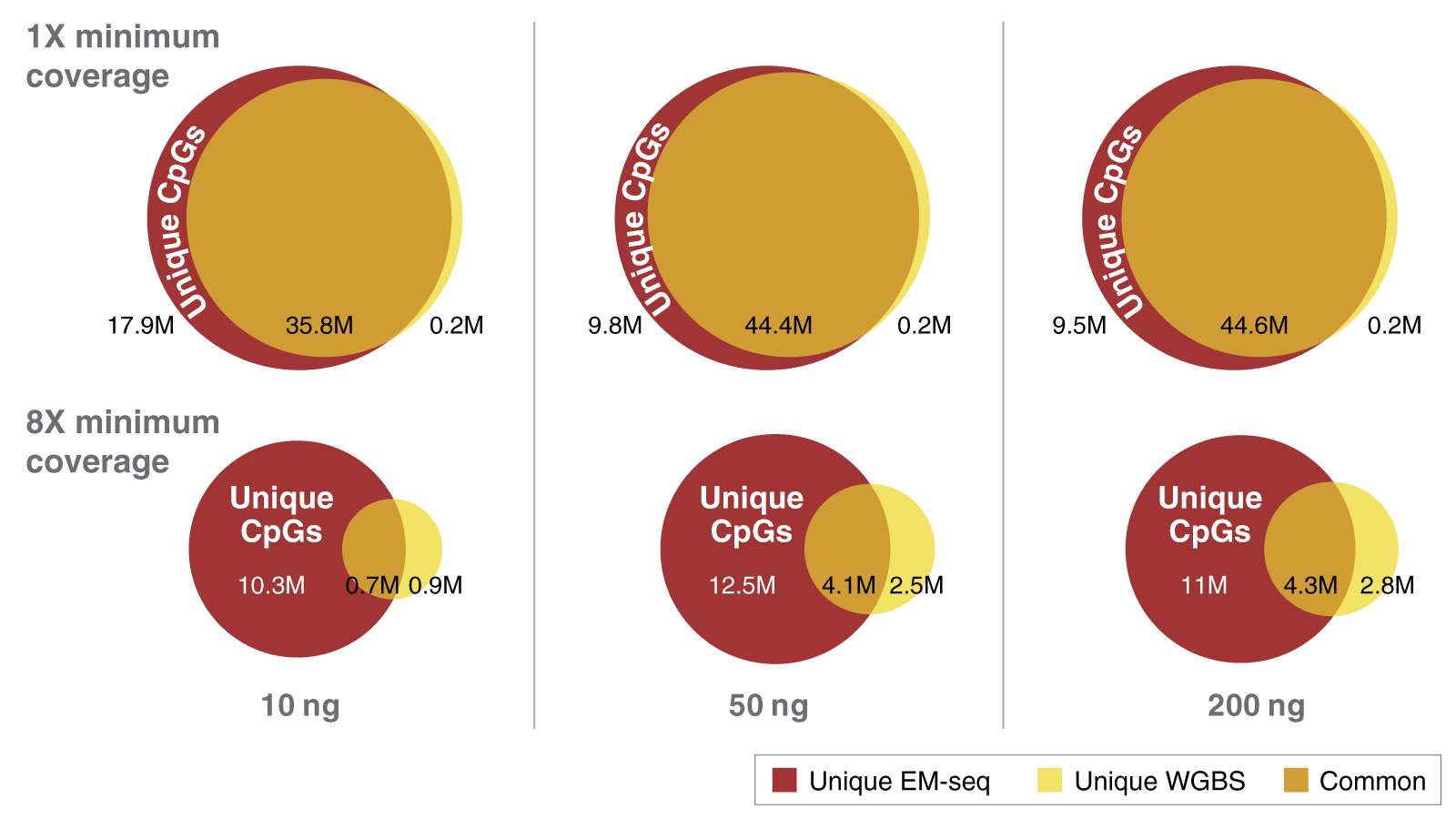
When both DNA strands are considered, there are approximately 56 million CpGs in the human genome. Complete detection of these CpGs is important. As you can see in the figure above, in combination with NEB’s highly efficient Ultra II library prep, EM-seq delivers superior detection of CpGs. The minimum coverages of unique CpGs with EM-seq compared to WGBS at various inputs is striking. EM-seq detects more CpGs at greater depth than WGBS using the same number of raw reads. This is particularly evident with lower DNA inputs. EM-seq is superior to WGBS and using sequencing reads from libraries generated with a 10 ng DNA input at a 1x coverage depth. WGBS detects 36 million CpG’s compared to 54 million for EM-seq. If a more stringent 8x coverage depth is required, EM-seq detects 11 million CpGs while WGBS only detects 1.6 million CpGs. That's a major difference in unique CpG coverage.
One of the main reasons for the increase in CpG detection is the intact nature of enzymatically converted DNA which results in a better genome wide coverage, and this is reflected in the superior GC uniformity compared to WGBS libraries. You get more sequence reads for a particular section of a genome, which gives you greater confidence in the consensus sequence generated from all the reads. Furthermore, NEB scientists optimized EM-seq to detect DNA methylation at single-base resolution from 100 pg of DNA. The full technical note on EM-seq is available here.
These EM-seq library attributes contribute to the accurate and reproducible analysis of DNA methylation targets. Using newer technologies like EM-seq is becoming more essential to get the real biological picture as NGS technology transitions from research to clinical use.

EM-seq improves results in many methylation analysis applications
Whole genome sequencing
Whole genome bisulfite sequencing (WGBS) is widely used to study DNA methylation at single base resolution, but its accuracy is significantly limited by DNA damage. It's been terrific to see recent scientific works demonstrate how useful EM-seq is for generating this type of data.
Looking at the big picture with whole genomes it's been established that WGBS libraries that have adaptors ligated prior to bisulfite treatment have reduced mapping rates and skewed GC content representation as well as an under-representation of G- and C-containing dinucleotides and an over-representation of AA-, AT- and TA-containing dinucleotides when compared to a non-converted genome. This source of bias affecting DNA methylation data was reported by Olova et al. (2018) Genome Biology. Many shortcomings of the original method were overcome using Post Bisulfite Adaptor Tagging (PBAT) type libraries where adaptors are introduced after bisulfite conversion to improve library yields and genome coverage. However, the fundamental issues of DNA damage related to bisulfite treatment remain with these post-conversion libraries. There is also another useful method called TET-assisted pyridine borane sequencing (TAPS) that combines enzymatic (TET1) activity and a chemical (pyridine borane) reaction to identify DNA methylation. Like EM-seq, TAPS-based methods do not introduce the same DNA damage as bisulfite treatment. TAPS can also be modified to look at various other cytosine modifications. Just keep in mind that the TAPS methods require generating TET1 enzyme. You also need to switch to new analysis pipelines since TAPS reads modified cytosines directly.
Excitingly, several groups of investigators have recently reported supportive evidence that EM-seq can outperform WGBS on many levels. Library insert sizes are larger, GC bias plots are normalized compared to standard libraries, and even genome coverage results in more CpGs detected compared to WGBS. These metrics are part of the reason why Morrison J, et al. Evaluation of whole-genome DNA methylation sequencing library preparation protocols. (2021) Epigenet. Chromatin recommends EM-seq for whole genome DNA methylation sequencing, based on their data with with fresh–frozen human fallopian tubes tissue samples. The EM-Seq protocol also compared favorably to the bisulfite sequencing-based approaches analyzed using high quality DNA inputs from human cell lines, in Foox et al. The SEQC2 epigenomics quality control (EpiQC) study (2021) Genome Biology which presented a multi-platform assessment and cross-validated resource for epigenetics research from the FDA’s Epigenomics Quality Control Group. In almost all comparisons, EM-Seq libraries captured more CpG sites at equal or better coverage. Also, Suhua Feng et al. Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing (2020) Epigenetics & Chromatin suggested that EM-seq is a more accurate and reliable approach than WGBS to detect DNA methylation. Data from the report Yanan Han et al. Comparison of EM-seq and PBAT methylome library methods for low-input DNA (2021) Epigenetics, suggests that EM-seq performed better overall compared to post-bisulfite adaptor tagging in whole-genome methylation quantification of low input samples.
Each of these studies support EM-seq for whole genome sequencing. It's especially neat to see the variety of sample types used, from high-quality cell lines and tissues, to low-input ng level of DNA derived from cerebral spinal fluid. There are so many amazing avenues of epigenomics research.
EM-seq converted DNA has been used for longer amplicon sequencing using Pacific Biosystems (PACBIO®) powered by Single Molecule, Real-Time (SMRT® ) Sequencing technology. Longer reads are required for phased whole genome sequencing which identifies specific allele expression on maternal and paternal chromosomes for studying complex genetic traits. Bisulfite converted DNA is too damaged to be used for longer read technologies. Long read TAPS (lrTAPS) has also been described where converted DNA was sequenced using Oxford Nanopore Technologies® and PACBIO® - again with the current lack of a commercialized source of TET1 as a practical caveat.
Applying EM-seq conversion to a reduced representation method provides a wider coverage of MspI digested regions compared to that of bisulfite reduced representation because DNA is more intact after enzymatic conversion.
Intriguing application possibilities with EM-seq
DNA methylation microarrays in theory
EM-seq could be substituted for bisulfite-converted DNA for use in methylation microarrays without changing single-stranded DNA probe collections or downstream analysis pipelines. Bisulfite converted DNA is often used in microarrays to identify methylated cytosines in CpG islands, differentially methylated sites, enhancers, or transcription factor binding sites. TAPS methods can’t be directly substituted without designing new single-stranded DNA probes because TAPS identifies 5mCs directly. Here again switching to enzymatic conversion is a convenient way to improve how well your data represents in vivo methylation.
Opportunities for 5hmC detection
Variations on the EM-seq and TAPS methods, as well as ACE-seq can be used to probe 5hmC content. These methods are non-destructive and give an alternate route to gain knowledge about 5hmCs role in gene regulation. Established methods that can interrogate 5mC and 5hmC individually based on bisulfite sequencing still incur DNA damage just like traditional bisulfite sequencing.
Bisulfite-seq is no bargain

NEB will not rent, sell or otherwise transfer your data to a third party for monetary consideration. See our Privacy Policy for details. View our Community Guidelines.
Don’t miss out on our latest NEBinspired blog releases!
- Sign up to receive our e-newsletter
- Download your favorite feed reader and subscribe to our RSS feed
Be a part of NEBinspired! Submit your idea to have it featured in our blog.


