Single end reads:
Read 1 should be trimmed with AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC (note: other vendors use different sequences)
For complete oligo sequences check the NEBNext Small RNA Library Prep for Illumina manuals.
No left trimming of reads is needed since the sequencing primer sits down immediately before the insert.
We recommend using a simple program like flexbar [1] for single end trimming.

Paired end reads:
Read 1 should be trimmed with AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC (note: other vendors use different sequences)
For complete oligo sequences check the NEBNext Small RNA Library Prep for Illumina manuals.
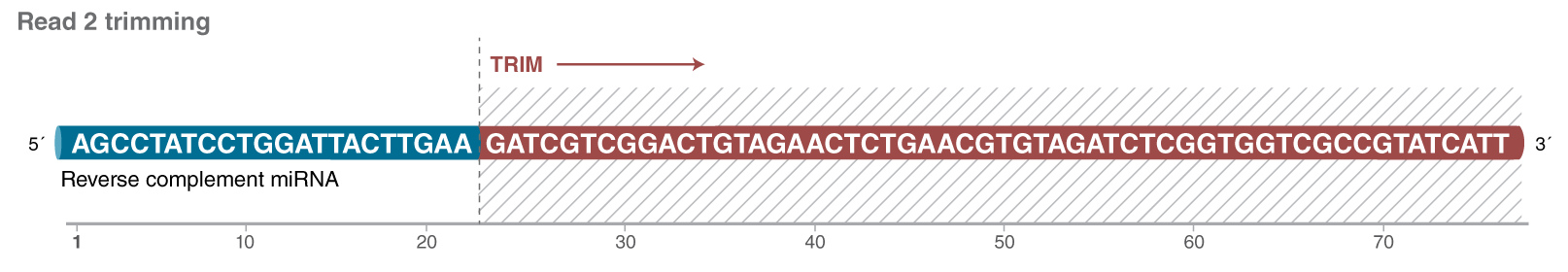
Read 2 (if present, not typically acquired with short inserts) should be trimmed with GATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATT
For paired end reads, presence of true adapter sequence requires that the insert is shorter than read length. In that scenario, both read 1 and read 2 contain information about the position of the adapter. Use of an adapter trimmer that takes advantage of this additional signal is advisable. For example Seqprep[2], Flexbar (-ap option)[1], or similar.
[1] Roehr, J. T., Dieterich, C., & Reinert, K. (2017). Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics, 33(18), 2941–2942.
https://doi.org/10.1093/bioinformatics/btx330
https://github.com/seqan/flexbar
