SNAP-tag® Technologies: Novel Tools to Study Protein Function
Scientists investigating the function of proteins in cells wrestle with rather basic questions: Where? When? With whom? Answering these questions for any given protein can be a challenging enterprise and, as most scientists are aware, there is often a painful discrepancy between the magnitude of the challenge and the tools available to address it.
New England Biolabs now introduces a set of protein tools for the specific labeling of fusion proteins with synthetic probes that will help scientists illuminate various aspects of protein function.
Covalent Labeling of Fusion Proteins Using SNAP-tag and CLIP-tag
The expression of a protein of interest as a fusion protein with an additional polypeptide (tag) that aids in the characterization of the protein was first exploited by the group of Jon Beckwith in 1980 (1): β-galactosidase was fused to the cytoplasmic membrane protein MalF to facilitate its purification. Since then, an ever increasing number of tags have become popular in various branches of biology. The practice of cell biology without the use of fluorescent proteins is unimaginable today, and the purification of recombinant proteins using affinity tags is standard practice.

However, despite the ubiquitous role of fusion proteins in protein science, traditional tags face two major limitations:
i. They are limited to properties that can be genetically encoded. For example, the spectroscopic properties of currently available fluorescent proteins are often inferior to synthetic fluorophores, but such fluorophores cannot be genetically encoded.
ii. Traditional tags are tailor-made for a specific application and a single tag is often not suited to the study of different facets of protein function: expression, transport, interaction and degradation.
An approach that addresses these shortcomings relies on tags that can be specifically labeled with synthetic probes such as fluorophores or affinity labels. NEB now introduces a suite of novel tagging technologies for specifically labeling fusion proteins in cell biology and protein science applications.
SNAP-tag
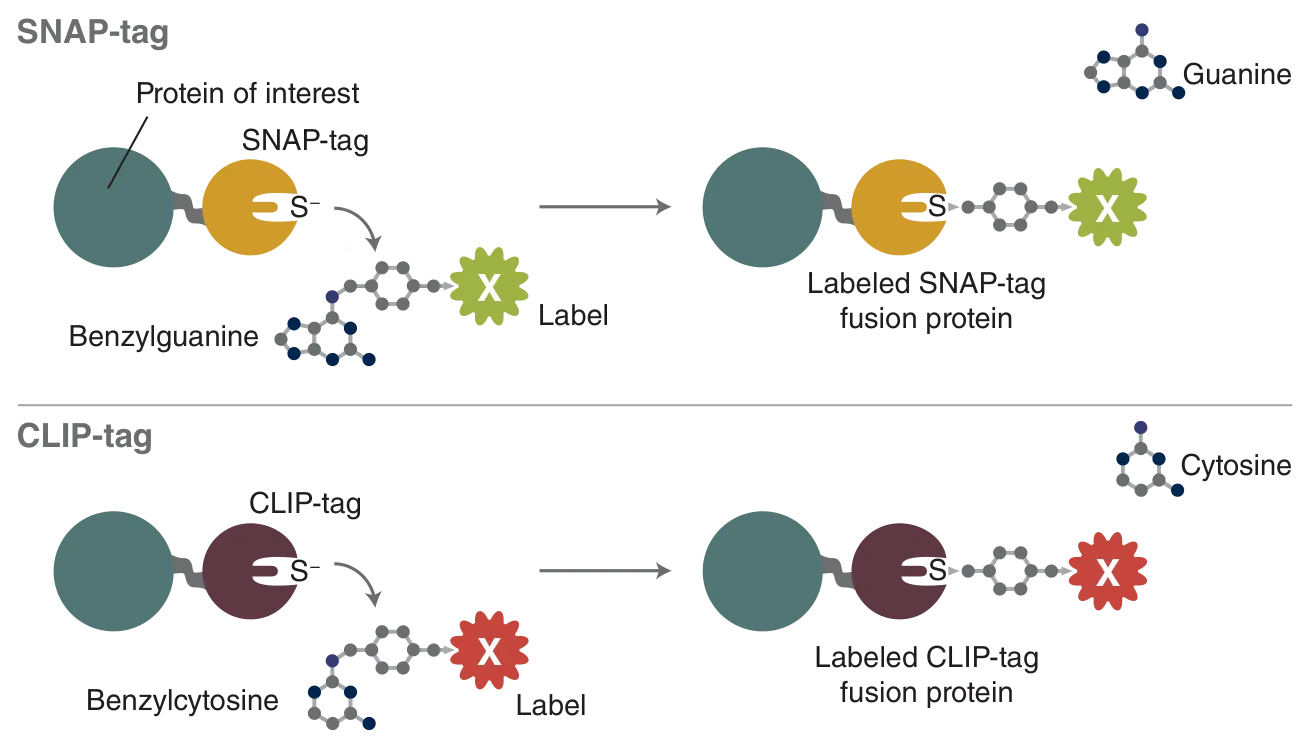
The most versatile of these tags is the SNAP-tag, a 20 kDa mutant of the DNA repair protein O6-alkylguanine-DNA alkyltransferase that reacts specifically and rapidly with benzylguanine (BG) derivatives (Figure 1), leading to irreversible covalent labeling of the SNAP-tag with a synthetic probe (2). SNAP-tag has a number of features that make it ideal for a variety of applications in protein labeling. The rate of the reaction of SNAP-tag with BG derivatives is to a large extent independent of the nature of the synthetic probe attached to BG, permitting the labeling of SNAP fusion proteins with a wide variety of synthetic probes.
Secondly, SNAP-tag has no restrictions with respect to cellular localization and expression host. Thirdly, SNAP-tag substrates are chemically inert towards other proteins, avoiding nonspecific labeling in cellular applications. Finally, many SNAP-tag substrates are cell permeable, permitting labeling of intracellular proteins in live cells.
CLIP-tag
The second tag introduced by NEB is a sibling of the SNAP-tag, the CLIP-tag (3). The CLIP-tag was created by engineering the substrate specificity of the SNAP-tag, permitting it to react specifically with O2-benzylcytosine (BC) derivatives (Figure 1). Since the SNAP- and CLIP-tags specifically react with orthogonal substrates, SNAP and CLIP fusion proteins can be labeled simultaneously and specifically with different synthetic probes in living cells. The main application of the CLIP-tag is dual-labeling of fusion proteins in conjunction with the SNAP-tag.

Application: Location, Timing and Interaction?
Protein Location
The proper localization and translocation of proteins is an integral aspect of cellular function. The labeling of SNAP and CLIP fusion proteins with synthetic fluorophores in living cells permits the determination of their cellular localization through fluorescence microscopy (Figure 2). As both the SNAP- and CLIP-tags can be labeled with different fluorophores, the simultaneous localization of more than one protein (via SNAP and CLIP fusions or in combination with fluorescent proteins) is possible. The labeling of SNAP and CLIP fusion proteins can be carried out either on fixed cells, as the tags retain their activity during fixation, or more commonly in living cells. Live cell imaging permits the dynamic, real-time study of protein translocation. A representative example is the monitoring of the nuclear redistribution of a SNAP fusion of the human estrogen receptor α (ER) upon incubation of cells with the partial ER antagonist 4-hydroxytamoxifen (6,7). Another very attractive feature of the labeling of fusion proteins is that the labeling itself can be restricted to certain locations of a cell. For example, use of cell-impermeable substrates of SNAP-tag, enable the fraction of a plasma membrane protein present on the surface of the cell to be visualized through specific labeling(8). The approach thus permits the discrimination of different populations of a cell surface protein: those properly translocated to the plasma membrane from those retained in the secretory pathway or already internalized, e.g., upon ligand binding. The possibility to restrict experiments to sub-populations of G protein-coupled receptors (GPCRs) that are properly folded and functionally presented cell surfaces has been exploited in a number of important studies (9,10). It should also be noted that such discrimination cannot be easily achieved when using fluorescent proteins.
Protein Timing
Since the timing of labeling of a given fusion protein is under experimental control, questions about protein trafficking, protein turnover, organelle dynamics and macromolecular assembly become open to investigation. The stability of the covalent labeling brings the possibility of using different dyes at different time points during the experiment to distinguish between older and newly synthesized proteins in living cells. Such multicolor pulse chase labeling experiments generate distinct populations of otherwise identical proteins with discriminating features determined only by the time point of the respective labeling of each population. This approach was exploited to investigate phospholamban synthesis and turnover over time: multicolor pulse chase labeling of this protein over a 24 hour period allowed the distinction of older and newer copies, uncovering a potential new role in myoblast differentiation (11). Jansen et al. labeled proteins translated within a defined window of the cell cycle to probe the role of centromeric protein A (CENP-A), a histone H3 variant, in centromere determination and cell division (12). Unexpectedly, these experiments revealed that CENP-A is inserted into centromeric histones uniquely in the G1 phase. Recently, McMurray and Thorner used multicolor pulse chase labeling to differentiate old and new molecules of two septins from budding yeast, Cdc10 and Cdc12, to probe their stability and recycling during dynamic structural transitions in cell division and development (13). The labeling experiments revealed that mechanisms governing septin incorporation are specific to each subunit and to the developmental state of the cell. It should also be noted that a sequential labeling with more than two fluorophores is possible; such labeling experiments are particularly attractive to study biological structure formation as was shown for cell wall synthesis in budding yeast (14). Finally, the non-overlapping substrate specificity of SNAP-tag and CLIP-tag permit simultaneous pulse-chase experiments to visualize different generations of two different proteins in one cell, further increasing the potential of the approach (3).
Protein Interaction
Protein-protein interactions are at the heart of most biological processes and the chemical labeling of fusion proteins also offers multiple opportunities to characterize such interactions. Proteins can be labeled with fluorophores ideally suited for fluorescence energy transfer experiments. This has been exploited to study the homo- and hetero-dimerization of GPCRs (9,10). In addition to providing the ability to select pairs of fluorophores that are better suited for FRET experiments than fluorescent proteins, chemical labeling brings the extra benefit that immature or improperly folded proteins retained in the secretory pathway do not contribute to the observed signal. Sophisticated FRET experiments can also be complemented with more traditional biochemical approaches: magnetic beads derivatized with the SNAP-tag substrate BG can be used for efficient pull-down experiments to identify and characterize interaction partners of the protein of interest. Finally, SNAP-tag fusions are also attractive for high-throughput assays of protein-protein interactions in vitro; either FRET-based, employing surface-plasmon resonance (15) or through the creation of protein microarrays (16,17). The possibility to address a single question through a variety of different experimental approaches makes the SNAP-tag particularly attractive for the analysis of protein-protein interactions.
The Future of SNAP- and CLIP-Tag protein?
The aforementioned examples from the literature demonstrate the potential of specific chemical labeling of fusion proteins, in particular the SNAP- and CLIP-tags, to address central questions in cell biology and protein science. Innovation in chemistry via the synthesis of new labeling substrates with advantageous properties opens up completely new ways to study protein function: the recently introduced small molecule fluorescent probes for live-cell super resolution microscopy (18) or the use of environmentally sensitive color-shifting dyes for ratiometric quantification (19) are good examples, or the use of biosensors for measuring call metabolites (20). The simplicity of the synthesis of such substrates and the availability of the necessary building blocks from NEB permit even those scientists with little background in chemistry to assemble their own substrates. Whereas advances in the development of new intrinsically fluorescent proteins force their users to go through continuous cycles of subcloning and characterization, users of the SNAP-tag and its relatives can be assured to directly benefit from such future inventions.
References
- Shuman, H.A., Silhavy, T.J., and Beckwith, J.R. (1980) J. Biol. Chem. 255, 168.
- Keppler, A., Gendreizig, S., Gronemeyer, T. et al. (2003) Nat. Biotechnol. 21, 86.
- Gautier, A., Juillerat, A., Heinis, C. et al. (2008) Chem. Biol. 15, 128.
- George, N., Pick, H., Vogel, H. et al. (2004) J. Am. Chem. Soc. 126, 8896.
- Yin, J., Straight, P.D., McLoughlin, S.M. et al. (2005) Proc. Natl. Acad. Sci. USA 102, 15815.
- Keppler, A., Kindermann, M., Gendreizig, S. et al. (2004) Methods 32, 437.
- Pick, H. Jankevics, H., and Vogel, H. (2007) J. of Mol. Biol. 374, 1213.
- Keppler, A., Pick, H., Arrivoli, C. et al. (2004) Proc. Natl. Acad. Sci. USA 101, 9955.
- Maurel, D., Comps-Agrar, L., Brock, C. et al. (2008) Nat. Methods 5, 561.
- Meyer, B.H., Segura, J.M., Martinez, K.L. et al. (2006) Proc. Natl. Acad. Sci. USA 103, 2138.
- Stenoien, D.L., Knyushko, T.V., Londono, M.P. et al. (2007) American Journal of Physiology 292, C2084.
- Jansen, L.E., Black,, B.E., Foltz, D.R. et al. (2007) J. of Cell Bio. 176, 795.
- McMurray, M.A. and Thorner, J. (2008) Curr. Biol, 18, 1203.
- Vivero-Pol, L., George, N., Krumm, H. et al. (2005) J. Am. Chem. Soc. 127, 12770.
- Kindermann, M., George, N., Johnsson, N. et al. (2003) J. Am. Chem. Soc. 125, 7810.
- Sielaff, I., Arnold, A., Godin,G. et al. (2006) Chembiochem 7, 194.
- Jongsma, M. A. and Litjens, R. H. (2006) Proteomics 6, 2650.
- Wang, L., Frei, M.S., Salim, A., Johnsson, K. (2019) J. Am. Chem. Soc. 141, 2770.
- Wang, L., Hibolt, J., Popp, C., Xue, L., Johnsson, K. (2020) Angew. Chem. Int. Ed. Engl. 59, 21880.
- Yu, Q., Pourmandi, N., Xue, L., Gondrand, Cc., et al. (2019) Nat. Matab. 1, 1219.
By Kai Johnsson, Ph.D., Ecole Polytechnique Fédérale de Lausanne
