Protocol for use with NEBNext Microbiome DNA Enrichment (E2612)
Protocols
Note: It is important to use sterile technique to avoid environmental DNA contamination.
DNA Preparation and Quantitation
Any method for the purification of protein-free genomic DNA can be used, including Proteinase K treatment followed by phenol/chloroform extraction and ethanol precipitation, lysozyme digestion, Qiagen® column preparation, NEB Monarch column preparation (for genomic DNA), or other methods. Sonication, nebulization, chaotropic salts, enzymatic fragmentation, rough handling, multiple freeze-thaws, or any other procedure that would cause DNA to shear, should be avoided. Samples should be in DNase-free TE buffer (pH 7.5) and be at least 15 kb or greater in size, and free of small molecular weight fragments. If the DNA fragment size is < 15 kb, enrichment will be less efficient. Determine DNA quality and quantity by agarose gel electrophoresis of the sample alongside a DNA marker (e.g., Lambda DNA-HindIII Digest, NEB #N3012). It is also important to quantitate
the amount of DNA in the experimental sample by A260 measurement using a
spectrophotometer, such as a Nanodrop® instrument or Qubit® Fluorometer.
Prebind MBD2-Fc Protein to Magnetic Beads
Before proceeding, you need to determine the volume of prepared beads required for enriching microbial DNA from your sample.- For every 6.25 ng of input DNA you will need 1 μl of MBD2-Fc-bound magnetic beads for enrichment; therefore, if your total input DNA is 1 μg, you will need 160 μl of MBD2-Fc-bound magnetic beads.
- The amount of MBD2-Fc-bound magnetic beads required (Y) can be calculated using the following equation:
Y = amount of MBD2-Fc- bound magnetic beads (μl) = Input DNA (ng)/6.25 ng/μl
For example:
| 1000 (ng) | = 160 μl |
| 6.25 ng/μl |
The following protocol will yield 160 μl MBD2-Fc-bound magnetic beads for a 1 μg input sample. For other input sample amounts, scale accordingly.
- Resuspend NEBNext Protein A Magnetic Beads by gently pipetting the slurry up and down until the suspension is homogeneous. Alternatively, rotate the tube on a rotating mixer gently for 15 minutes at 4°C. Do not vortex.
- Prepare 1X Bind/wash Buffer on ice by diluting 1 part NEBNext Bind/wash Buffer (5X) with 4 parts DNase-free water. One individual reaction, from start to finish, will require 4 ml of 1X Bind/wash Buffer.
- In one tube, add 16 μl of MBD2-Fc protein and 160 μl of Protein A Magnetic Beads. For input amounts other than 1 μg, add (0.1 x Y) μl of MBD2-Fc protein and (Y) μl of Protein A magnetic beads. Mix by pipetting up and down until the beads are completely homogeneous, at least 5-10 times.
- Mix the bead-protein mixture by placing the tube in a rotating mixer for 10 minutes at room temperature.
- Briefly spin the tube and place on the magnetic rack for 2–5 minutes or until the beads have collected to the wall of the tube and the solution is clear.
- Carefully remove the supernatant with a pipette without disturbing the beads.
- Add 1 ml of 1X Bind/wash Buffer (kept on ice) to the tube to wash the beads. Pipette up and down until the beads are completely homogeneous, at least 5-10 times.
- Mix the beads on a rotating mixer for 3 minutes at room temperature.
- Briefly spin the tube and place on the magnetic rack for 2–5 minutes or until the beads have collected to the wall of the tube and the solution is clear.
- Carefully remove the supernatant with a pipette without disturbing the beads.
- Repeat steps 7–10.
- Remove the tube from the rack and add 160 μl of 1X Bind/wash Buffer (kept on ice) to resuspend the beads. For input amounts other than 1 μg, add (Y) μl of 1X Bind/wash Buffer to resuspend the beads. Mix by pipetting up and down a few times.
- The MBD2-Fc-bound magnetic beads are stable for up to 7 days at 4°C.
Capture Methylated Host DNA
- Add 1 μg in up to 200 μl input DNA to the tube containing the 160 μl of MBD2-Fc-bound magnetic beads. For other DNA input amounts, add DNA to (Y) μl of MBD2-Fc bound magnetic beads.
- Add undiluted Bind/wash Buffer (5X) for a final concentration of 1X. (For example add 10 μl of Bind/wash Buffer (5X) if the DNA input sample was 40 μl, add 4 μl of Bind/wash Buffer (5X) if the DNA sample was 16 μl). Pipette the sample up and down until the beads are completely homogenous, at least 5-10 times.
Volume of 5X Bind/wash Buffer to add (μl) = Vol input DNA (μl)/4
- Agitate the tube on a rotating mixer for 15 minutes at room temperature with rotation.
Collect Enriched Microbial DNA
- Briefly spin the tube and place on the magnetic rack for 5 minutes until the beads have collected to the wall of the tube and the solution is clear.
- Carefully remove the supernatant with a pipette, without disturbing the beads and transfer it to a clean microcentrifuge tube. This supernatant contains the target microbial DNA. Store this sample at –20°C or proceed directly to purification:
Purify the samples by either AMPure XP bead clean-up or ethanol precipitation.
Option A: Agencourt® AMPure® XP/SPRIselect Bead Cleanup
- Vortex AMPure XP/SPRIselect Beads to resuspend.
- If your enriched sample volume x 2.8 from the previous step exceeds your tube volume, split into two tubes.
- Add 1.8X volume of resuspended AMPure XP or SPRIselect beads to the sample. Mix well by pipetting up and down at least 10 times. Be careful to expel all of the liquid out of the tip during the last mix.
- Incubate samples for at least 5 minutes at room temperature.
- Place the tube on the appropriate magnetic stand to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic stand.
- After 5 minutes (or when the solution is clear), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
- Add 400 μl of freshly prepared 80% ethanol to the tube/plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
- Repeat Step 7 once for a total of two washes. Be sure to remove all visible liquid after the second wash. If necessary, briefly spin the tube/plate, place back on the magnetic stand and remove traces of ethanol with a p10 pipette tip.
- Air dry the beads for up to 5 minutes while the tube/plate is on the magnetic stand with the lid open.
Caution: Do not over-dry the beads. This may result in lower recovery of DNA. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn brown and start to crack, they are too dry.
- Remove the tube from the magnetic stand. Elute the DNA target from the beads by adding 50 μl of 1X TE (use 25 μl for each tube if sample was split in two).
- Mix well by pipetting up and down 10 times. Incubate for at least 2 minutes at room temperature. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing back on the magnetic stand.
- Place the tube/plate on the magnetic stand. After 5 minutes (or when the solution is clear), transfer eluate to a new microcentrifuge tube (combine the eluates if sample was split in two).
- The sample can now be used for NGS library construction or other downstream analysis.
Option B: Ethanol Precipitation
- Add 2.5 volumes of 100% ethanol*, incubate for 10 minutes on ice, then centrifuge the sample for 30 minutes at 13,000 rpm (16,000 rcf). Remove the ethanol, allow the pellet to air dry, and then resuspend the pellet in a small quantity (50 μl) of TE buffer.
- If the ethanol pellet contained any residual beads, the resuspended sample can be placed on the magnetic rack 5 minutes to concentrate the beads on the inner wall of the tube, and the supernatant can be transferred to a fresh microcentrifuge tube.
- The sample can now be used for NGS library construction or other downstream analysis.
For ethanol precipitation of the proteinase 1 < digested host DNA also add 0.3 M (final) Na-Acetate
NGS Library Construction
Enriched samples are compatible with NEBNext DNA Library Prep reagents for the following platforms:
Optional Protocol for Eluting Captured Host DNA
This step elutes the captured host DNA from the MBD2-Fc-bound magnetic beads.
- While the tube with the beads (section "Collect Enriched Microbial DNA", Step 2) is still in the magnetic rack, add 1 ml of 1X Bind/wash Buffer (kept on ice) to wash the beads.
- Carefully remove the wash buffer with a pipette without disturbing the beads.
- Add 150 μl of 1X TE and 15 μl of Proteinase K to the sample pellet. Mix beads by gentle vortexing (1,200 rpm) or by flicking the tube. (Note: The pellet may be difficult to resuspend initially due to the high concentration of genomic DNA bound to the beads.) Incubate the slurry in a heat block or thermomixer set at 65°C for 20 minutes, with occasional mixing (3–5 times).
- Briefly centrifuge the sample at 13,000 rpm in a microcentrifuge.
- Place the tube on the magnetic rack for 2–5 minutes or until the beads have collected to the wall of the tube and the solution is clear.
- Carefully remove the supernatant and transfer to a fresh microcentrifuge tube.
- This eluted sample contains the methylated host DNA. Store this sample at –20°C, or proceed directly to purification via using AMPure XP bead cleanup or ethanol precipitation.
Downstream Analysis
Quantitation of DNA by agarose gel electrophoresis
- Prepare the unenriched sample by diluting 0.1 μg of unenriched input DNA in TE Buffer for a final volume of 20 μl.
- Aliquot 20 μl of the unenriched DNA, supernatant fraction containing the enriched mircobial DNA, and eluted fraction containing the eukaryotic host DNA into separate tubes. Add 5 μl of Gel Loading Dye, Blue (6X) (NEB #B7021) or other loading buffer to each sample.
- Load 20 μl of each sample alongside an appropriate DNA marker (λ DNA-HindIII Digest, NEB #N3012) on a 0.8% agarose gel. Since the supernatant fraction containing enriched microbial DNA will be depleted of host DNA, the total amount of DNA seen on the gel will be reduced compared to the input control.
Validation of enrichment by qPCR
- Prepare the unenriched input sample by diluting the same quantity as previously used for enrichment in TE Buffer in a final volume of 50 µl.
- For each sample (input, enriched, and purified microbial DNA, aliquoted and purified host (human DNA), aliquot the volume appropriate for your qPCR reaction in triplicate. Do this for all primer pairs used (16s rRNA . gene and RPL30 gene or other primers for host DNA).
- Add the appropriate volume of qPCR primers for your qPCR kit. Dilute as needed. Primers are supplied as 20 µM each.
- Perform qPCR and analyze results as appropriate for your qPCR reagents and instrument.
- In the microbial fraction, little to no change in the Cq value for enriched universal bacterial control primers should be observed between the input fractions. A significant Cq shift should be observed between the input and enriched fraction with RPL30 or other host primers. In the host fraction, a small shift of Cq should be observed with the RPL30 or other host primers. A significant shift should be observed for the 16SrRNA primers.
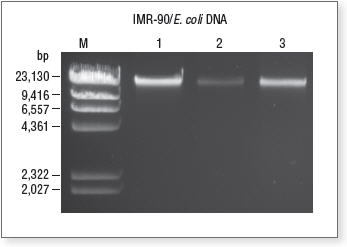
Agarose gel containing unenriched, enriched and captured host DNA
Lane M, Lambda DNA-Hind III Digest, NEB #N3012; Lane 1, Unenriched input DNA; Lane 2, Supernatant fraction containing enriched microbial DNA; Lane 3, Eluted DNA from magnetic bead pellet containing host DNA.

