Solutions for the Expression of Difficult Proteins
Script
Speaker 1:
Good day everyone. Thanks for holding, and welcome to the New England BioLabs Solutions for the Expression of Difficult Proteins conference call. Just a quick reminder, today's call is being recorded. Now at this time, I would like to turn things over to our host, Dr. Jim Samuelson. Dr. Samuelson, please go ahead sir.
Dr. J Samuelson:
Yes. Welcome to everyone who has chosen to join us for this information session on solutions for the expression of difficult proteins. I will start by outlining the most common problems encountered during the expression of recombinant proteins in E. coli and specific solutions will be given for each problem. Following me will be Corinna Tuckey, who will describe the utility and advantages of the PURExpress in vitro protein synthesis kit. And then Mehmet Berkman will highlight the SHuffle line of E. coli strains for producing proteins with complex disulphide bonds.
The two most common problems during recombinant protein expression are low expression level and solubility of the target protein. This is particularly true when expressing eukaryotic genes in bacterial hosts. When expressing membrane proteins, researchers must assume from the start that the protein will be difficult to express in functional form. If a membrane protein expresses at a high level, almost certainly some of this protein will be found in the insoluble fraction. As a solution NEB offers a tunable T7 expression strain for the production of membrane proteins and other difficult targets so that the level of functional protein may be optimized.
Regardless of the type of protein, isolating a homogeneous sample is often challenging. In today's presentation I will describe a new strain that we have engineered to aid the isolation of pure his-tag proteins. And finally, many proteins from higher organisms require complex disulfide bonds for stability and proper folding. NEB also offers solutions for expressing these types of proteins, either in vitro or in vivo.
First let me address the problem of low target protein expression. Since many of today's expression vectors encode Shine-Dalgarno sequences it is easy to overlook problems with translation initiation. When designing expression constructs, make sure that the vector does indeed encode a properly spaced ribosome binding site and also analyze for secondary structure at the five prime end of the messenger RNA. This analysis may be accomplished with software programs such as Mfold, UNAfold and other similar programs.
If you find that the five prime UTR is not optimized for expression then simply change the vector, the ribosome binding site, or adding an affinity tag at the N-terminus often enhances expression. However, expression problems are most often due to open reading frames with non-optimal codons for E. coli. As a solution, various expression strains are available that offer higher levels of so-called "rare tRNAs". But, the best solution today may be to completely redesign the gene.
This is now very economical as many companies offer this service. A gene can be ordered, it is optimized for E. coli expression and it can be at your lab within three weeks.
Finally, low expression may be due to uncontrolled expression of toxic protein during the culture outgrowth phase. This can result in loss of cell viability or loss of the expression plasmid. Maintenance of the expression plasmid should always be checked by plating cells at the point of induction on drug selection plates versus non-drug plates. And if the number of colonies on the drug plate is less than fifty percent of the number on the non-drug plate, steps need to be taken to stabilize the host vector system.
If you're using a T7 system, by far the best way to control basal expression is by adding T7 lysozyme. The T7 lysozyme is a natural inhibitor of T7 RNA polymerase activity.
Now the next problem we'll address is insoluability of the target protein. Folding heterologous protein within E. coli is far from an exact science. Therefore the cause of mis-folding is often unknown. It is thought that folding problems are primarily due to the inherent nature of each unique protein and the capacity of the chaperone pool to mediate its proper folding. When confronted with insolubility problems, the first resort should be to lower the temperature to fifteen to twenty degrees during the induction period. This simple solution helps in many, but not all situations. Lowering the inducer concentration may also be necessary, remember less is more. Less expression will often result in more protein produced in the desired form. Second option is to use an N-terminal fusion partner. Many proteins have physiological binding partners and if the physiological partner cannot be co-expressed then the fusion partner may help to solubilize the target. Maltose binding protein displays chaperoone-like activity and it enhances expression as well as solubility. Furthermore, MBP acts as an affinity module for purification on amylose resin.
However, if you do not wish to use a fusion partner and you're using a T7 system, then solubility may be improved by using a tunable T7 host strain such as Lemo21(DE3). Lemo21(DE3) is a BL21(DE3) strain where expression can be carefully controlled. Lemo means less is more. This strain was developed by Xbrane Bioscience at Stockholm university and NEB is the only supplier for this strain as a competent cell. With B21(DE3) expression can be so robust that all of the target goes into inclusion bodies. However, with the Lemo21 strain, activity of the T7 RNA polymerase can be adjustable. By moderating expression, cellular chaperones then have a better chance to fold the target protein.
You see, in this slide we have a diagram of how the Lemo system works. On the left side of the slide is pLemo which expresses T7 lysozyme from the rhamnose promoter. pLemo is compatible with almost all common T7 expression vectors. The system works by expressing lysozyme at different levels until you find a level that is appropriate for your particular protein.
This slide shows an example where Lemo21 was used to increase the level of soluble target protein. Brugia malayi proteins are notoriously difficult to produce in E. coli as shown on the left side of this slide where most of the target protein is found in the insoluble fraction when expressed in BL21(DE3). However, when expressed in the Lemo strain more target protein becomes soluble as the level of Lys-Y inhibitor is increased by increasing the level of rhamnose in the culture.
So in this case, less is more. Less T7 expression equals more soluble protein. The Lemo21 strain is also especially useful for the expression of membrane proteins. In E. coli alpha-helical membrane proteins get inserted into the intermembrane by the SecYEG translocase, shown here in this slide. In addition, most periplasmic and outer membrane proteins travel through the same protein conducting channels. Therefore, the Sec translocase is recognized as the major bottleneck when a heterologous membrane protein is over expressed in the cell. If the SRP-Sec pathway becomes jammed by over expression of the target then the host cell will simply stop growing. So to avoid this problem and to avoid loss of expression, membrane protein expression should be finely tuned to a level that is tolerated. That's where the Lemo21 strain is so useful. As shown in this slide, optimization of expression may be accomplished in one day using one strain. In this example, a polytopic membrane protein was fused to GFP so that the expression level could be easily measured.
Researchers at Xbrane Bioscience found that one thousand micromolar rhamnose was the optimal level for this particular protein. At that level, greater than 20,000 units of fluorescence were observed. In contrast, when rhamnose is not added to the culture to moderate expression, less that 2000 fluorescence units are observed. Although not shown here, when the same construct was expressed in BL21(DE3) only 1400 units were observed.
The next difficulty that we all have encountered is the isolation of sufficiently pure protein. Today most researchers turn to affinity purification for isolating their target. So NEB offers three primary tools for affinity purification. The first tool is the pMAL Protein Fusion and Purification System. In this system, the target is fused to MBP, it's expressed then purified on amylose resin, and if necessary the target is cleaved away from the tag using protease.
The next tool is the Impact System. In this system the target protein is fused to an intein and to a chitin binding tag. The fusion is bound to chitin and the target protein is eluted by intein-mediated cleavage and the tag and intein stay behind on the column. Therefore, proteases are not necessary to liberate the target protein when using the Impact System.
Although NEB does not offer nickel or cobalt resin, we are the only company that offers a strain designed for improving the purity of his-tag proteins and that strain is NiCo21(De3). NiCo21(DE3) is a BL21 derivative where we've gone in an altered the host chromosome to tag three metal binding proteins with the chitin binding domain tag. Furthermore, the GlmS gene is mutated so that the glmS protein no longer binds to nickel or cobalt resin.
These chromosomal alterations were made since all four of these contaminating proteins are important for cell viability, so the respective genes could not simply be deleted. This slide outlines the purification process when NiCo21 is used as the host. The first step is a standard metal chelate column and you see in the middle of the slide is a typical eltution profile where you have the abundant target protein but also some contaminating proteins from E. coli. So then the next step is to use a chitin column to clear away the CBD-tagged contaminating proteins.
In this example, we show a real world example of using the NiCo21 strain to purify a his-tag protein and we compare the process directly to a process where BL21(DE3) was used. In the elution fraction from BL21(DE3) you see that there's several contaminating proteins. The three most prominent contaminants are indicated by filled symbols. On top we have a filled square corresponding to RNA which is 74 kDa. And the next protein down is glmS which is 67 kDa. And then the lower fuzzy band is SlyD which runs between 20 and 30 kDA. Whereas in the NiCo elution fraction, that is elution from the nickel column, glmS protein is not present, indicating that the mutated glmS was not able to bind initially to the nickel column. The highest contaminant that is indicated is RNA-CBD and you see that it's shifted compared to wild-type RNA since the CBD tag is approximately 7 kDA. And then down further is SlyD-CBD, again the mobility is shifted up due to the CBD tag.
And finally when this elution from the NICo strain is passed over a chitin column, the CBD tag proteins are sufficiently removed. And the net result is the target protein is present at 97% purity.
These purity numbers come from an analysis using the Caliper Lab Chip GXII system.
The next two problems are often difficult to distinguish. When several bands light up on a Western Blot, this could mean that the target is subject to proteolysis or this could also mean that the bands correspond to internal translation products.
In this example, since the lower band is so prominent, it appear that the lower band corresponds to an internal translation product. Internal translation initiation may occur when expressing archaeal or eukaryotic genes since these genes often have internal ATG or GTG codons with upstream purine sequences that can act as ribosomal binding sites. So the solution is simple. Mutate these problem regions while maintaining the conserved codons if possible. This mutagenesis can be done by site-directed mutagenesis or again, the gene could be submitted for redesign and gene synthesis by an outside company.
If you find that it's clear that the target is being degraded by proteases, be certain to use a protease-deficient strain. One of the protease-deficient strains as the host. These are BL21, Lemo21, NiCo21, or any of the T7 express strains are also protease-deficient, as they are B strains and they lack ompT and Lon protease activity. Since protealysis can be a sign of poor folding, you can adjust the culture conditions or change host strains to try to improve folding and therefore reduce proteolysis.
Other solutions are obvious such as adding protease inhibitors during processing and if you're expressing a multi-domain protein or a fusion protein, do not introduce unnecessary arginine or lysine residues within linker regions during mutagenesis or clone design. And also, if all of those solutions fail, the target can be expressed in vitro with the PURExpress system which is essentially protease-free, unlike first generation S30 translation systems.
And that brings us to the final problem for discussion and that is expression of proteins with complex disulfide bonds. NEB offers two solutions. The first solution is the PURExpress in vitro protein synthesis kit which can be used with a disulfide bond enhancer component. And our cell-based solution is to express these types of proteins in one of the SHuffle strains of E. coli which have been engineered to facilitate folding of proteins with non-consecutive disulfide bonds. These two solutions will be described in more detail by my colleagues, Corinna Tuckey and Mehmet Berkman. And the next segment will be presented by Corinna.
Thank you for your attention everyone.
Corrina Tuckey:
Hello everyone, my name is Corinna Tuckey and I am the current product manager for the PURExpress in vitro protein synthesis kits that are offered by New England BioLabs.
The first slide that I want to show you today has to do with in vitro systems with defined components and very common systems with defined components such as DNA amplification, or PCR, and RNA amplification such as in vitro transcription are widely used today, and allow us to have more control and customization in these complex processes. These systems contain anywhere from eight to fourteen molecules, macromolecules, and small molecules. Traditional in vitro protein synthesis is done from cell extracts. The New England BioLabs now offers the PURExpress system which is a reconstituted in vitro transcription translation system from E. coli. This system is more defined and controllable for protein translation.
My next slide shows a simplified schematic of the NEB PURExpress system and what I want to point out here is that it is very easy to use. If you look at the left hand side of the slide, you can see that it essentially consists of two solutions: solution A and solution B. These solutions contain amino acids, tRNAs, NTPs, T7 RNA polymerase ribosomes, translation factors, and tRNA synthetases, just to name a few items that have been reconstituted into our system.
All you need to do is add your template under the control of a promoter for the T7 RNA polymerase at the correct ribosome binding site in order to get synthesis of your protein of interest. On the right hand side of the slide you can see the polysome actively synthesizing protein. This reaction takes about two hours and you should have your protein of interest.
So the general features of PURExpress and other cell-free translation systems are that protein expression is decoupled from host cell viability and this allows you to explore greater sequence space in studying protein function and design. One can create modified proteins for the study of functional domains or to investigate critical residues in protein function, and you can also create hybrid proteins such as designer proteins for therapeutics and similar studies like that. Also, the feature of being able to incorporate non-natural amino acids allows for greater functionality in protein sequence space. Due to the defined nature of PURExpress, it makes it particularly well suited for studies beyond the traditional cell-free systems. Its essential nuclease and protease-free nature allow you to add a linear DNA template such as PCR products that might be necessary for higher throughput methods. An example of this might be in an enzyme evolution study.
It also allows for a more robust recovery of the activity-encoding nucleic acid template that has been introduced into this system. Then the last point I want to make here, and a very convenient point for PURExpress is that you can produce analytical amounts of protein for direct testing right from the reaction, and these tests can involve things like protein-protein interactions and testing for enzymatic activity.
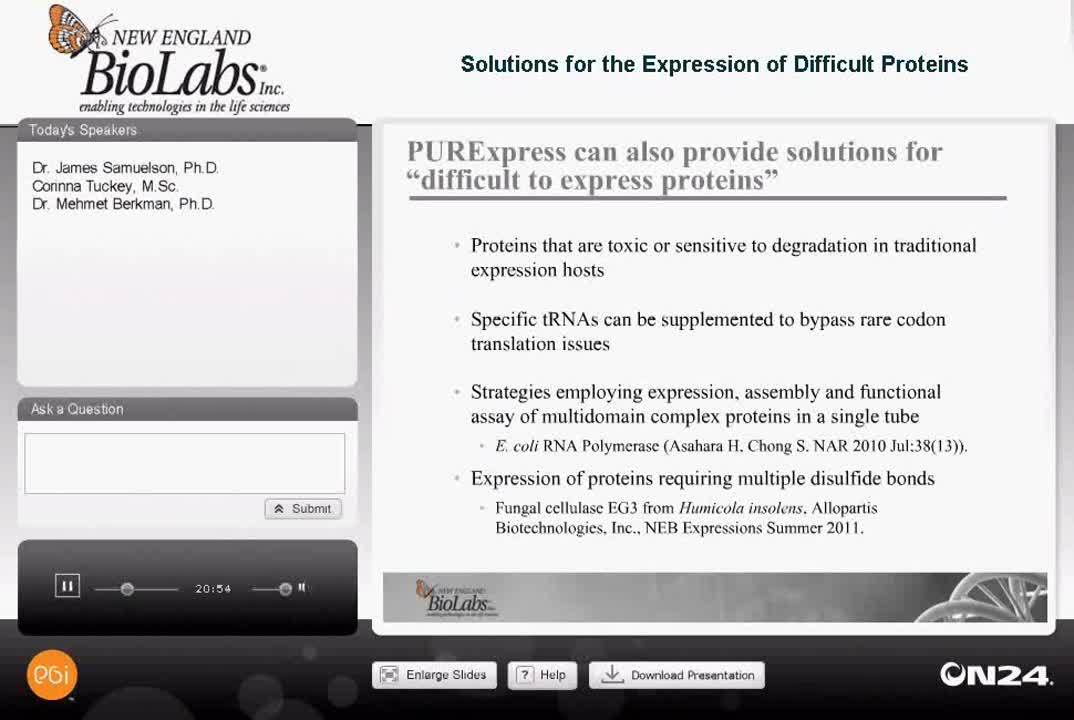
PURExpress is also excellent for providing solutions for quote "difficult to express" proteins and as Dr. Samuelson mentioned earlier, proteins that are toxic to cells are particularly well-suited for in vitro expression, or proteins that are sensitive to degradation in traditional expression hosts.
Also, with codon issues you can specifically add tRNAse that will supplement the tRNAse available and bypass rare codon translation issues. Two other topics for difficult to express proteins that I'm going to go into further detail about today are the expression and functional assay of a multi-domain complex protein. This study was done by one of the research labs at New England BioLabs, it was published about a year ago in Nucleic Acids Research and the citation is on this slide. This was a study involving the E. coli RNA polymerase holoenzyme.
The other topic that I'm going to go into more detail about is the expression of proteins requiring multiple disulfide bonds, and these can be non-consecutive bonds. This study was done by Allopartis Biotechnologies in San Francisco and will be published in the NEB Expressions, summer edition. We also have an application out describing these studies further and this was done with the fungal cellulase EGIII from Humicola insolens.
So the in vitro expression assembly and functional assay of E. coli RNA polymerase was done where with this strategy that coupled expression and assembly of multiple genes with functional detection in a single tube. The researchers used only DNA templates instead of purified proteins to do this study and the paper that they published describes the synthesis assembly of the functional E. coli RNA polymerase holoenzyme with the expression of a reporter gene, the firefly luciferase.
They then went on further to use this system to demonstrate sigma factor-dependent promoter-specific transcription initiation.
My next slide shows a picture of the E. coli RNA polymerase holoenzyme that is also complexed with the four-junction DNA. The enzyme core is shown as molecular surfaces in the subunits of alpha-I, alpha-II, and omega are shown in gray. The beta subunit is shown in blue and the beta prime subunit is shown in pink. We also have the associated sigma factor shown as a backbone with the alpha helices shown off to the right and the DNA four junctions shown in green with the minus thirty-five and minus ten elements shown in yellow.
My next slide is a very busy slide. Just to illustrate what was put into the reaction tube for this experiment. Essentially, it's quite simple. The researchers took all of the subunits for the E. coli RNA polymerase, the alpha, beta, beta prime, omega, and the sigma factor, put them under the control of the T7 RNA polymerase promoter. They expressed these in vitro with the PURExpress system. And it's shown in the bottom left-hand corner. The E. Coli RNA polymerase core enzyme associated with the sigma factor to form an active holoenzyme which then read the firefly luciferase reporter gene off of specifically the E. coli RNA polymerase promoter, and we were able to ascertain a read out from the luciferase as a function of formed E. coli RNA polymerase.
Our next figure is a figure from the paper and it is basically showing the detection of the E. coli RNA polymerase directly as read by firefly luciferase activity. Firefly luciferase activity is shown in the y-axis and the first bar farthest to the left is what activity we saw when we added all subunits of the RNA polymerase to the reaction with the sigma factor. The next three bars show that you get no firefly activity if you omit the alpha, beta, or beta prime subunit from the reaction. This also indicates that there was no background activity of E. coli RNA polymerase in the reaction.
If you omit the omega subunit from the reaction, you still do see the E. coli RNA polymerase activity and this may be that it's not required for the function of the polymerase. There are some other discussion points around this that can be found in the paper.
Further confirmation of each subunit was confirmed by immunoblot and this is shown in this figure where antibodies against the alpha, beta, beta-prime and the sigma32 factor were shown that these subunits were definitely expressed in the in vitro reaction.
As the summary to this part of the talk, I'd just like to illustrate that the in vitro synthesis and assembly of functional E. Coli RNA polymerase was demonstrated. Six proteins with molecular weights ranging from 10 to a 155 kDA were expressed in a single tube. And this strategy allows for the immediate detection of the activity of RNA polymerase and eliminated the need for the purification of the RNA polymerase and then a separate in vitro transcription reaction.
I'm now going to switch to the problem of proteins with disulfide bonds. We offer a PURExpress disulfide bond enhancer. This reagent can be added to a PURExpress reaction and what it does is it improves the ability of PURExpress, or S30 lysate if that's what you're using, to produce correctly folded proteins that contain extensive disulfide bond patterns.
It works by assisting with the oxidation of the cysteine thiols and correcting mis-oxidized substrates. It can increase the yield of soluble and functionally active protein. The protein in this example, as mentioned before is EGIII from the fungi H. insolens. It's an endoglucanase that creates internal glucan chain scissions.
My next slide shows the disulfide bonds that are contained in EGIII. The left-hand panel is illustrating the two non-consecutive disulfide bonds that are found in the carbohydrate binding module of this enzyme. In the right panel you can see that there is also a disulfide bond in the catalytic core and that they're all shown in yellow in these ribbon structures. Again, I just want to repeat here that this work was done in conjunction with Allopartis Technologies in San Francisco.
My next slide shows the effect of the disulfide bond enhancer supplementation in the PURExpress reaction on the activity relative to 250 ng of the in vivo expressed EGIII. The y-axis is showing micromole glucose equivalents liberated in an assay done directly again on the PURExpress reaction for enzyme. The x-axis is showing that the disulfide bond additives one and two were titrated in the reaction to get the optimal amount to add for best expression and activity of the protein, and the first bar shows that if you do not add the disulfide bond enhancer to the reaction, you get a little activity but not as much as if you add one microliter of the first component and four microliters of the second component, and that gave the best activity. And again, just to mention it, this was compared relative to 250 ng of the SHuffle-expressed purified EGIII.
The summary to this section: the active EGII was successfully expressed using PURExpress in the in vitro synthesis kit and the addition of the disulfide bond enhancer increased the enzyme activity thirteen-fold. The endoglucanase activity was measured directly from the PURExpress reactions and active EGIII was also successfully expressed in and purified from SHuffle-competent E. coli to milligram quantities.
My colleague, Dr. Berkman is now going to talk more about Shuffle-competent E. coli.
Mehmet Berkman:
Hello everyone, my name is Mehmet Berkman and I'd like to continue our discussion on solutions to proteins that are difficult to fold by introducing you a novel E. coli strain engineered here at NEB, designed to express proteins that require disulfide bonds for their folding.
The story of protein folding doesn't stop after synthesis. There are many proteins that require covalent post-translational modifications. This could be proteolytic cleavage of their pro-domain for certain secreted proteins, or covalent modifications which attach sugars, methyl groups, fatty acids, or in the case we're going to talk about today, certain proteins may form disulfide bonds. I'd like to remind everyone that these events in a living cell are highly regulated both in space and in time.
So let me begin from the very beginning and remind everyone what a disulfide bond is. A disulfide bond occurs by the oxidation of the thiol groups within a cysteine, here represented as the SH and forming a covalent bond. And this reaction occurs in the presence of an electron acceptor. You could readily reverse the reaction in the presence of an electron donor, thus making disulfide bonds uniquely sensitive to their environment.
Disulfide bonded proteins are not as rare as one might think. For example, in a recent bioinformatics study, a third of the human proteins were shown to be localized to the endoplasmic reticulum where disulfide bond forming machinery exists and half of those proteins were predicted to form disulfide bonds. Since disulfide bonds greatly increase the stability of a protein, they're usually found in secreted proteins or proteins that carry information from one cell to another, such as antibodies or scavenging enzymes that take nutrition from the environment, proteins that are associated with the membrane that are facing the environment all are enriched in disulfide bonds.
Not surprisingly, a recent study looking into the therapeutic proteins produced globally showed that all the therapeutic proteins either are represented exclusively or partially by disulfide bonded proteins. However, making disulfide bonded proteins in E. coli is not easy. Whether these are either secreted to the periplasm where oxidation occurs, or you need to use specialized E. coli cells such as the Shuffle cell I'm going to tell you about today. Even so, these are not guaranteed to solve the problem and one might need to further engineer the protein, the strain, or express chaperones to get correctly folded proteins.
So it is therefore quite essential that we have new and versatile expression tools to address any protein folding issue.
So how did we engineer this? Here's a brief schematic showing you how the Shuffle cells work. In blue you can see the oxidizing periplasm which is much smaller in volume and limited in energy, because it has no ATP and chaperones compared to the reducing cytoplasm.
However, the cytoplasm is reducing and will not tolerate disulfide bonds. What we have done is genetically engineered the strain to have a diminished reducing potential, such that the cytoplasm is now both oxidizing and reducing and we have co-expressed an isomerase chaperone which further increases the fidelity of disulfide bonded proteins.
Let me begin by showing you how disulfide bonds form in a normal wild type E. coli. As I said, the cytoplasm is a reducing environment and that is because there are numerous reductases such as thioredoxins that will catalyze the reduction of any disulfide bonds formed. They receive their reducing potential from NADPH and if any disulfide bond may form in the cytoplasm, it will quickly be enzymatically reduced to its reduced form. A protein that requires disulfide bonds needs to escape this compartment which is usually done through a signal peptide that targets it to a machine such as the Sec apparatus that will bring it to the periplasm where an enzyme that we have named DsbA will quickly catalyze the formation of disulfide bonds. This process is very fast and is believed to occur during translocation into the periplasm and thus it should have a protein that is complex patterned and non-consecutive disulfide bonds, more often than not DsbA will mis-oxidize an mis-fold the protein.
When the protein is misfolded, another enzyme called DsbC recognizes this misfolded protein and through a process that we don't truly understand, isomerizes it to its correct native for and will make the protein active. Now, DsbC is critical to the property of Shuffle cells, so with your permission I'm going to go a little more in depth explanation into the properties of DsbC.
DsbC is a homodimer periplasmic protein, having two major domains: a thioredoxin domain, here shown in blue, and a dimerization domain, here shown in gray. The dimerization domain brings the two thioredoxin folds together, resulting in this cleft that is believed to be hydrophobic. The busy end of the activity, the isomerase activity is catalyzed by the redox-active cysteines which face this cleft. We believe this hydrophobic cleft is essential doe DsbC to distinguish between misfolded and correctly folded proteins, as misfolded proteins tend to have their hydrophobic core residues exposed.
Here the crystal structure of DsbC we can see those two domains better. You can see in white the thioredoxin domain brought together through a linker to the dimerization domain and the cleft. If we do a surface film model of DsbC, we can clearly see that cleft and if we look at the charges should be shown DsbC, we see that the cleft is very rich in hydrophobic, or uncharged, amino acids. Thus, allowing it to interact with almost any protein.
Furthermore, if you take a protein such as GAPDH, which has no disulfide bonds, and denture it in vitro and in the test tube ad DsbC, you can reconstitute it back to its folded state quite efficiently using DsbC. So DsbC has quite a good chaperone property. Much better than its functional eukaryotic homologue PDI. Here you see that PDI is a not as efficient chaperone as DsbC is. And if you add to this reaction a mutant of DsbC that is only monomeric, that does not have the cleft or the hydrophobic patch, you see that its chaperone property is diminished.
Also, if you look at all the proteins that have been published in literature that have been expressed in the periplasm of E. coli and look at their activity or yield when expressed in the absence of DsbC, you see that some proteins represent as a percentage are quite active in the absence of DsbC shown to be one hundred percent active while others are not. So there seems to be kind of a dependency on whether a protein's correctly oxidized or not by DsbA. And if you take a closer look, what you will notice is that all the proteins that fold correctly in the absence of DsbC seem to have their disulfide bond pattern in a consecutive manner. We believe this supports the notion that disulfide bonds occur during translocation into the periplasm, however, in the SHuffle strain, we make disulfide bonds in the cytoplasm. So whether this correlation exists in the cytoplasm is not shown and I will show you some data that suggests that it might not be. So, not only is the property of your substrate protein quite important, its connectivity is also, needs to be considered when looking into methods of producing proteins.
So, a little brief description of how we genetically engineered SHuffle to allow disulfide bond formation to occur in the cytoplasm. As I said, NADPH is the reducing pool of the cell, that's where most of the electrons are stored, and NADPH donates its electrons to a set of enzymes. One is the glutaredoxin pathway represented here by Gor. Gor takes those electrons from an NADPH and transfers them through a cascade through several proteins through, at the end, several reductases that are called glutaredoxins. These reductases, these glutaredoxins will catalyze the reduction of any disulfide bonds, making sure the proteins do not have the stable disulfide bonds. There's the secondary pathway, represented by the thioredoxins, which take electrons from TrxB, transfer it so finally to thioredoxins which do the same thing. These two pathways are essential for maintaining the protein in their reduced state. What we have done is knocked out, genetically removed those two genes Gor and TrxB and when we do this the cells are no more viable. They will die as certain enzymes are required to maintain their reduced state for viability.
We then select for supressors that restore viability to these dead cells. And once we investigated to map those supressors, we found that it mapped to another pathway that is the peroxidase pathway. AhpC is another reductase, well it's a peroxidase, that scavenges hydrogen peroxide away from the cytoplasm and the mutant version of AhpC which was a supressor of a lethal strain, lost its peroxidase activity and gained a reductase activity. It was then shown that AhpC star, the reduction version cannot reduce glutareadoxin and allow cells to become viable again. While the thioredoxins remain in their oxidized state and catalyze the oxidation of proteins. So in the SHuffle cells we have this confusing state where both the reduction and the oxidational disulfide bonds are occurring. And whether your protein gets oxidized or not, we believe it's governed by the laws of thermodynamics and folding of each unique protein.
But what we have additionally done is co-express at high levels the naturally periplasmic protein DsbC in the cytoplasm. This greatly increases the fidelity of the disulfide bond forming machinery, thus allowing for expression of multi complex disulfide bonded proteins.
So we wanted to check the idea whether the expression of a protein in the cytoplasm is better than in the periplasm. Here we take the example of a galactosidase which has only one disulfide bond and when we express this in the cytoplasm of wild type cells you can see that the extracts of that cell cannot cut its sugar substrates, and we see no activity of this protein when expressed in the cytoplasm of wild type cells. If you then target the same construct to the periplasm, you see now they are capable of cutting their substrate and you get less than 2000 units of activity per grams of cell. It's the same construct that's now expressed in the cytoplasm of SHuffle that get much higher units per gram cell, indicating at least in the case of this protein, that you could get a lot more protein in the cytoplasm of SHuffle, than in the periplasm of wild type cells. Although this hasn't been shown for a vast number of proteins, we do believe this can tend to be true for most proteins.
We then had the hypothesis in mind, we said that the DsbC should increase the fidelity, but the disulfide bonds from machinery in the cytoplasm is much different than the periplasm so we wanted to test that idea, and what we did was we took three proteins and expressed it in cells, in cytoplasm in oxidizing cells that either have a DsbC or not. The dark bar represent cells that do not have DsbC and the light gray bar represents cells that do. So in the case of the luciferase which has multiple cysteines with unknown pattern, because it hasn't been crystallized yet, we see that DsbC actually is detrimental to its folding and we see a reduction activity when expressed in cells that express DsbC. Another protein, urokinase, which is highly dependent on DsbC expression in the periplasm, does not seem to have much dependency, and we see a modest improvement in activity. While another protein, tissue plasminogen activator, so protease. This protein can only be expressed in SHuffle cells and not in oxidizing cytoplasmic strains.
So whether your protein is required DsbC will depend on your protein, but we believe that the general tendency will be DsbC will be helpful as it has an isomerase and a chaperone property.
While we constructed these strains, we did two versions of the SHuffle strains. One we engineered based on the parent E. coli B strain, known for its quality as a good protein expression strain and the classic E. coli K12 strain. And while we were doing research we noticed that our B cells were performing better than our K12. So we took the same three proteins and expressed them in SHuffle B, which we at NEB call SHuffle express or SHuffle K12 sub. Just SHuffle. And what we saw in the case of luciferase was that it increased activity in SHuffle B cells. We saw the same thing for urokinase and the same thing for vtPA. So this indicates that somehow, at least in our hands, SHuffle B cells out perform SHuffle K12. I would not say this is true for every protein, I'm sure there are exceptions, but in our hands so far we always encourage people to try SHuffle B first if they're going to try one version over the other.
A lot of the questions I get regarding the SHuffle strain is "I have a protein, how do I express it? What should I try?" So for people who are researchers on just your average lab bench, we thought we should do the protein expression optimization in the same conditions. For companies that have high throughput expression profiles, they probably know how to express their protein, but your average researcher usually has three parameters that they modify: temperature; growth phase induction, "do I induce it early or late?"; and "how much do I induce it?" Those are the three most commonly manipulated parameters. So we thought we should take a test number of proteins and run them through these three parameters and find out if there's any obvious pattern or any correlation. And so we would express at various temperatures, take the optimum temperature and then express it at various growth phases, early, mid or late, and take the optimum temperature and the optimum expression phase and then try various concentrations of IPTG to induce our substrate. And using the optimum conditions that we found we would purify our protein and try to understand yield and specific activities.
So we did this for a panel of proteins and here's the summary of all of them. And you can see that certain proteins have complex disulfide monomers and others don't. And here's a summary of the most optimum conditions. What I'd like to bring your attention to is that the final yields of proteins, they dramatically depending on the protein. For vtPA, which folds poorly, we get negligible amounts, whereas for our cellulase you get tremendous amounts of protein. And the cellulase here was actually expressed by our collaborators at Allopartis.
We also noticed that a novel auto-expression method using Invitrogen's Magic Media greatly improved the final yield. We did not understand the exact mechanism of this, but when we expressed the same construct by just letting them grow overnight in this Magic Media supplied by Invitrogen, we had a six-fold improvement in activity for a very poor folding protein. So, perhaps for SHuffle strains we recommend trying the Invitrogen Magic Media as an improvement method.
We also noticed that the majority of the proteins folded better at lower temperatures with only a few of them folding at 37 degrees and we've since noticed that SHuffle strains behave better at lower degrees, so we always recommend that these cells are treated at lower temperatures and expression should be, if you're going to try out different temperatures, try out lower temperatures first.
Let me just summarize what I just said. We think the cytoplasm of SHuffles are a better folding environment than the periplasm. We think the SHuffle B versions of the cell lines are better than the K12. We recommend that the SHuffle cells be treated with some gentility and only grown at thirty-degrees and try lower temperatures when expressing your protein of interest. Although I didn't show you the data today, DsbC is in its correct redox state in the cytoplasm and highly over expressed and in most cases it is helpful, it is not essential. And the final yields of your protein will depend on your protein itself.
With that I'd like to sum up our presentation today. Please visit our web page or call us for any technical assistance regarding any of the products we mentioned today. Here is a quick list of the products and I will leave this on later for the viewers to watch. Again, I said please feel free to visit our webpage or even call us, and the data that was presented today wasn't just our work, but a collaboration of numerous scientists, both at New England BioLabs and our partners at Allopartis and Xbrane Biosciences. All of them are here individually thanked and with that I'd like to thank you for your attention and we would like to take any questions should there be any. Thank you very much.
Speaker 1:
Thank you. And ladies and gentlemen, at this time if you do have any questions for our panel, simply type them into the question box and click submit.
Speaker 1:
And again ladies and gentlemen, any questions or comments at this time, simply type them into the question box and click submit to get any of your questions or comments to our presenters.
Ladies and gentlemen it looks like we haven't any questions this afternoon.
And again ladies and gentlemen, looks like we have no questions today so we'd like to go ahead and conclude today's New England BioLabs conference call. We'd like to thank you all so much for joining us today and wish you all a great afternoon. Goodbye.

