Improving Enzymatic DNA Fragmentation for Next Generation Sequencing Library Construction
Introduction
The Human Genome Project (HGP), which was officially completed in 2003, was considered to be one of the world’s largest collaborative projects of its time (1). This involved many research groups worldwide and had the lofty goal of deciphering all 3 billion bases of the human genome. The project cost almost $4 billion dollars and took 13 years to complete with the available technology. Over a decade later, advancements in next generation sequencing (NGS) technologies have enabled sequencing of a human genome to become routine, taking less than two days, and at a tiny fraction of the cost of the original HGP.
The ability to quickly and inexpensively sequence whole genomes has truly revolutionized genomics research. Where once single genes or families of genes were studied, now whole genomes, exomes, transcriptomes and epigenomes are interrogated. With recent advances, such as the ability to multiplex and sequence many samples at once, NGS has transitioned from a basic research tool into the clinic, where it impacts discovery, diagnostics and treatment of disease.
Advances in genomics driven by NGS, as well as advances in the technology itself, continue at an amazing pace and move us closer to the realization of personalized medicine, where clinical decisions are tailored to an individual’s genome. However, if this pace is to continue, advances in all aspects of the technology must also continue. This includes early steps of the sequencing workflow, specifically in the preparation of samples, before they are sequenced.
To date, there are no sequencing platforms that can sequence intact DNA. Therefore, prior to sequencing, DNA molecules must be fragmented, or broken, into smaller pieces. These DNA fragments are then converted into libraries, by different methods depending on the sequencing platform to be used (Figure 1). In all cases, the libraries generated consist of the fragments of the unknown DNA to be sequenced, flanked by pieces of known DNA (adaptors), which are specific to each sequencing platform.
Figure 1: Traditional library preparation workflow

DNA Fragmentation Approaches
One of the major bottlenecks to sample prep is the first step: DNA fragmentation. The size of the DNA fragments generated depend on the sequencing platform being used, and can range from several hundred base pairs for short read sequencing technologies (e.g., Illumina®, Ion Torrent™) to >10 kb pieces for long read sequencing technologies (e.g., Pacific Biosciences® and Oxford Nanopore Technologies®). Methods for fragmenting DNA are broadly split into two basic categories: mechanical and enzyme-based. Mechanical shearing methods include acoustic shearing, hydrodynamic shearing and nebulization, while enzyme-based methods include transposons, restriction enzymes and nicking enzymes. Although many different options exist to fragment DNA, final fragment size, amount of starting material, upfront capital investment, and scalability must be considered when choosing a fragmentation method. Critically, in order to be useful for NGS, the method used must shear the DNA sufficiently randomly, so that the library being sequenced is fully representative of the original sample.

Mechanical Shearing
Options for mechanical fragmentation of DNA range from small plastic nebulizer devices to sophisticated electronic instruments. The most commonly used technique utilizes focused acoustic shearing devices, such as the instruments made by Covaris®. This involves focused transmission of high-frequency, short wavelength acoustic energy on the DNA sample. The size of the DNA fragments generated (150 – 5,000 bp) is controlled by changing both the intensity and the duration of the acoustic waves, and the protocols used are the same regardless of the amount or GC content of the DNA. Cost, challenges of scalability and sample loss (often caused by sample transfer after shearing), are some of the reasons that users of this method seek alternatives, especially as throughput increases.
If larger DNA fragments are required, hydrodynamic shearing can be used. In this method, hydrodynamic shear forces are applied by pushing DNA through the small orifice of a syringe. Size is controlled by altering the speed at which the DNA is pushed through the syringe. Centrifugation can also be used to create hydrodynamic force, by pulling the DNA sample through a hole with a defined size. Here, the rate of centrifugation determines the degree of DNA fragmentation. DNA fragments generated with hydrodynamic shear forces are typically in the range of 1-75 kb, but require large DNA input amounts (> 1 µg) and throughput is low. Nebulization is another method used to mechanically fragment DNA. Nebulization uses compressed air to force DNA through a small hole in a nebulizer unit and DNA fragment size is determined by the pressure used. Although this method is inexpensive and fragment size is somewhat tunable (typically 700 – 5000 bp in size), microgram quantities of DNA are required for starting material, and the method is most suitable for small numbers of samples.
Enzymatic Fragmentation
Enzyme-based fragmentation of DNA is an attractive alternative to mechanical shearing methods, as it does not require upfront capital expense, is amenable to quickly processing many samples at the same time, and reduces sample loss. Historically, the main concern with this method has been sequence bias, as many enzymes that cleave DNA have recognition sequences or sequence preferences.
Transposases fragment DNA by cleaving and inserting a short double-stranded oligonucleotide to the ends of the newly cleaved molecule. The inserted oligonucleotide must contain a sequence that is specific to the particular transposase being used. While this method is fast and has low input requirements, the known sequence bias associated with transposases make them incompatible with some applications.
Improving DNA Fragmentation for NGS Library Construction
To address the challenges associated with existing fragmentation approaches, NEB has developed a fragmentation system, the NEBNext® Ultra™ II FS DNA Library Prep Kit (NEB #E7805, E6177), in which a unique enzymatic fragmentation reagent is fully integrated into library preparation to generate low bias, high quality NGS libraries, with a simple, scalable workflow (for more information see page 6).
In order to reduce the NGS sample prep bottleneck, improvements in both performance and ease of use were necessary. In this work, we have focused on the DNA fragmentation step. Our new DNA fragmentation reagent is combined with end repair and dA-tailing reagents, and subsequent adaptor ligation is also carried out in the same vial (Figure 2). For low input samples, PCR amplification is performed prior to sequencing.
Figure 2: NEBNext Ultra II FS Kit workflow

Performance
Increased Library Yields
The use of enzymatic fragmentation can result in higher library yields as compared to mechanical shearing workflows, as the latter results in sample loss and DNA damage. Achieving sufficient library yields for high quality sequencing from very low input amounts can be especially challenging with mechanical shearing of DNA, a situation compounded by the preference to amplify libraries using as few PCR cycles as possible. Integration of our unique fragmentation reagent with end repair and dA-tailing, removing sample cleanup prior to ligation and eliminating multiple transfer steps all help to minimize sample loss. When combined with the high reaction efficiences of each step in the workflow and lack of DNA damage cuased by mechanical shearing, NEBNext Ultra II FS generates higher yields than library preparation using mechanical shearing methods. High library yields can be achieved with input amounts as low as 100 pg of human genomic DNA with amplification, or as low as 50 ng for PCR-free workflows (Figure 3, page 3).
Figure 3: NEBNext Ultra II FS DNA produces higher yields of PCR-free libraries
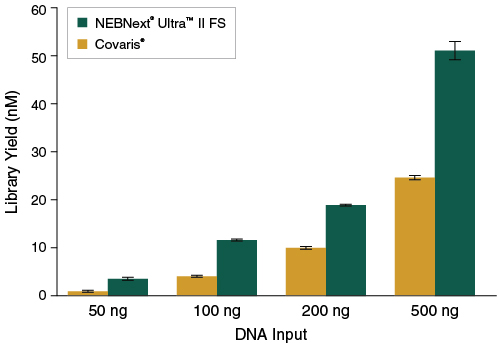
Libraries were prepared from Human NA19240 genomic DNA using the input amounts shown, without amplification. NEBNext Ultra II FS libraries were prepared using a 20-minute fragmentation time. “Covaris” libraries were prepared by shearing each input amount in 1X TE Buffer to ~200 bp using a Covaris instrument, followed by library construction using the NEBNext Ultra II DNA Library Prep Kit (NEB #E7645). Library yields were determined by qPCR using the NEBNext Library Quant Kit for Illumina (NEB #E7630). Error bars indicate standard deviation for an average of 2 libraries.
Figure 4: NEBNext Ultra II FS DNA provides uniform GC coverage with human DNA over a broad range of input amounts
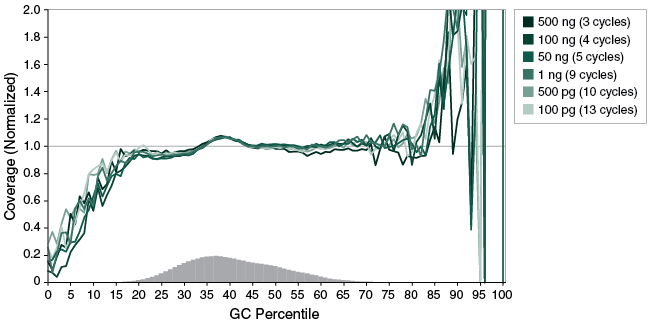
Libraries were prepared from Human NA19240 genomic DNA using the input amounts and number of PCR cycles shown, and a 20-minute fragmentation time was used. Libraries were sequenced (2 x 76 bp) on an Illumina® MiSeq®. Reads were mapped to the hg19 reference genome using Bowtie 2.2.4 and GC coverage information was calculated using Picard’s CollectGCBiasMetrics (v1.117). Expected normalized coverage of 1.0 is indicated by the horizontal grey line, the number of 100 bp regions at each GC% is indicated by the vertical grey bars, and the colored lines represent the normalized coverage for each library.
Figure 5: NEBNext Ultra II FS DNA provides uniform GC coverage for microbial DNA over a broad range of GC composition
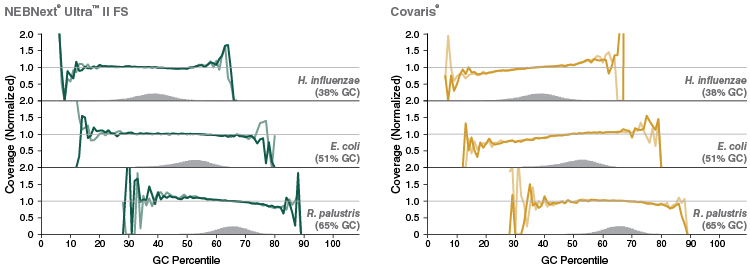
Libraries were prepared using 1 ng of a mix of genomic DNA samples from Haemophilus influenzae, Escherichia coli (K-12 MG1655) and Rhodopseudomonas palustris, with 9 PCR cycles, and sequenced on an Illumina MiSeq. NEBNext Ultra II FS libraries were prepared using a 20-minute fragmentation time. “Covaris” libraries were prepared by shearing 1 ng of DNA in 1X TE Buffer to an insert size of ~200 bp using a Covaris instrument, followed by library construction using the NEBNext Ultra II DNA Library Prep Kit (NEB #E7645). Reads were mapped using Bowtie 2.2.4 and GC coverage information was calculated using Picard’s CollectGCBiasMetrics (v1.117). Expected normalized coverage of 1.0 is indicated by the horizontal grey line, the number of 100 bp regions at each GC% is indicated by the vertical grey bars, and the colored lines represent the normalized coverage for each library.
Figure 6: NEBNext Ultra II FS DNA libraries show reduced markers of oxidative damage compared to libraries produced by mechanical shearing
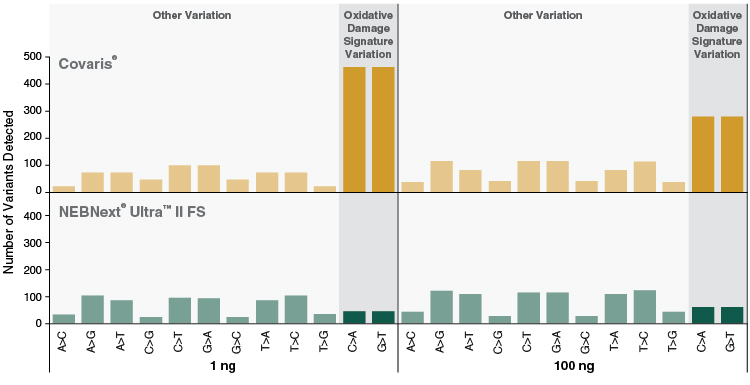
Libraries were prepared from 1 ng and 100 ng Human NA19240 genomic DNA, using 9 and 4 PCR cycles, respectively. NEBNext Ultra II FS libraries were prepared using a 20-minute fragmentation time. “Covaris” libraries were prepared by shearing each input amount in 1X TE Buffer to ~200 bp using a Covaris instrument, followed by library construction using the NEBNext Ultra II DNA Library Prep Kit (NEB #E7645). Libraries were sequenced on an Illumina® HiSeq® 2500 (2 x 75 bp). 723M reads were randomly sampled (seqtk) and aligned to the GRCh38 full reference genome using bwa (0.7.15). Adaptors were trimmed prior to alignment using trimadap (r9). Duplicates were marked using samblaster (0.1.24). Variants were called on chromosome 1 using freebayes (1.0.2.29) with frequency based options requiring at least 10 reads per site. More variants are observed for C>A and G>T transversions compared with all other variants for PCR-amplified Covaris libraries. These variants indicative of oxidative damage are not pronounced in NEBNext Ultra II FS libraries.
Ease of Use
Robustness of DNA Fragmentation
Consistent and reliable fragmentation is critical for a new method to be adopted. We optimized this new fragmentation system to be insensitive to variables such as input amounts, GC content, and DNA buffer conditions. In practice, these details are often unknown for a sample, requiring clean up and quantification prior to traditional enzymatic DNA fragmentation methods. Even when all of the variables are known, traditional enzymatic methods require different fragmentation parameters for each type of sample and DNA input amount. This new fragmentation system addresses all of these issues by requiring just a single-fragmentation protocol for the full range of input amounts (100 pg – 0.5 μg) (Figure 7, page 5) and for the full range of GC content (Figure 5). Also, input DNA can be in water, Tris, 0.1X TE or 1X TE (Figure 8, page 5). Fragmentation using the new system is time dependent, and final library sizes ranging from 100 bp – 1 kb can be generated by simply changing the fragmentation time.
Figure 7: NEBNext Ultra II FS DNA provides consistent fragmentation regardless of input amount
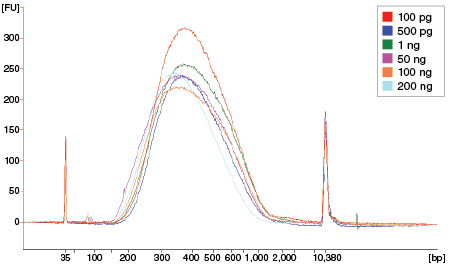
Libraries were prepared from Human NA19240 genomic DNA using the input amounts shown. NEBNext Ultra II FS libraries were prepared using a 20-minute fragmentation time. Library size was assessed using the Agilent® Bioanalyzer®. Low input (1 ng and below) libraries were loaded on the Bioanalyzer without a dilution. High input libraries were loaded with a 1:5 dilution in 0.1X TE.
Figure 8: NEBNext Ultra II FS DNA provides consistent fragmentation of DNA in water, Tris or TE
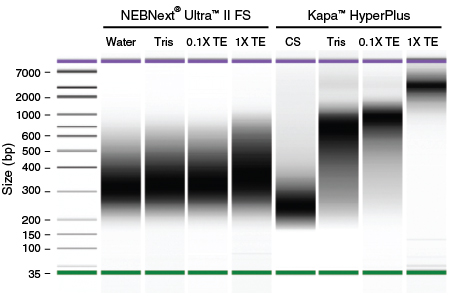
Libraries were made using 100 ng Human NA19240 genomic DNA using the NEBNext Ultra II FS kit or the Kapa HyperPlus Kit. Fragmentation conditions targeting ~200 bp inserts were used, which would generate ~320 bp libraries. For the NEBNext Ultra II FS kit, input DNA was in H2O, Tris, 0.1X TE or 1X TE . For the Kapa HyperPlus kit, libraries were made using the recommended dilution of the supplied Conditioning Solution (CS), or using DNA in Tris, 0.1X TE or 1X TE, in the absence of either Conditioning Solution or 3X bead clean up. Library size distribution was assessed using the Agilent Bioanalyzer. Fragmentation is consistent for the NEBNext Ultra II FS kit for DNA in H2O, Tris, 0.1X TE or 1X TE.
Conclusion
The continued expansion of the use of next generation sequencing depends in significant part on overcoming the limitations and bottlenecks associated with high-quality library preparation, including the initial DNA fragmentation step. While acoustic shearing has for some time been the method of choice for NGS, limitations in terms of instrumentation, throughput and sample damage necessitate sourcing an alternative solution for many uses. This new method for enzymatic DNA fragmentation and library preparation addresses these issues, further streamlines the process and improves the quality of NGS libraries. The broadening of the input amount range to as low as 100 picograms enables access to high-quality sequencing of samples not achievable by previous methods, and the reliability and ease of use of the method not only allows automation, but also successful adoption by users with a wide range of laboratory skills.
View a PDF of this feature article
References
- https://www.battelle.org/docs/default-source/misc/battelle2011-misc-economic-impact-human-genome-project.pdf
- M. Costello et al. (2013) Nucleic Acids Research, 41, e67.
- L. Chen, et al. (2017) Science, 17, 355: 752-756.

