Protocol (E6000) for use with End User Supplied Primers and Adaptors
Protocol
Starting Material: 1–5 μg
of Fragmented DNA to 200 bEnd Repair of Fragmented DNA
- Mix the following components in a sterile microfuge tube:
Fragmented DNA 1-75 μl Phosphorylation Reaction Buffer (10X) 10 μl T4 DNA Polymerase 5 μl Deoxynucleotide Solution Mix 4 μl T4 Polynucleotide Kinase 5 μl DNA Polymerase I, Large (Klenow) 1 μl Sterile H2O variable Total volume 100 μl - Incubate in a thermal cycler for 30 minutes at 20°C.
- Vortex AMPure XP beads to resuspend.
- Add 160 μl (1.6X) of resuspended AMPure XP beads to the ligation reaction. Mix thoroughly on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of 80% freshly prepared ethanol to the tube/pcr plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target by adding 40 μl sterile water to the beads. Mix well on a vortex mixer or by pipetting up and down, and put the tube/pcr plate in the magnetic stand until the solution is clear.
- Without disturbing the bead pellet, carefully transfer 32 μl of the supernatant to a fresh, sterile microfuge tube.
Alternatively, adaptor ligated DNA can be purified on one purification column. Purify DNA sample on one QIAquick column and elute in 32 μl of sterile water or elution buffer.
- Mix the following components in a sterile microfuge tube:
End Repaired, Blunt DNA 32 μl NEBuffer 2 (10X) 5 μl Deoxyadenosine 5'-Triphosphate 10 μl Klenow Fragment (3' → 5' exo-) 3 μl Total volume 50 μl - Incubate in a thermal cycler for 30 minutes at 37°C.
- Vortex AMPure XP beads to resuspend.
- Add 90 μl (1.8X) of resuspended AMPure XP beads to the ligation reaction. Mix thoroughly on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of 80% freshly prepared ethanol to the tube/pcr plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target by adding 15 μl sterile water to the beads. Mix well on a vortex mixer or by pipetting up and down, and put the tube/pcr plate in the magnetic stand until the solution is clear.
- Without disturbing the bead pellet, carefully transfer 10 μl of the supernatant to a fresh, sterile microfuge tube.
Alternatively, adaptor ligated DNA can be purified on one purification column. Purify DNA sample on one MinElute® (Qiagen) column and elute in 10 μl of sterile water or elution buffer.
- Mix the following components in a sterile microfuge tube:
*Adaptors are not provided; please use adaptors appropriate to the specific application. If necessary adjust the adaptor concentration to a final adaptor to DNA molar ratio of 10:1.dA-Tailed DNa 10 μl Quick Ligation Reaction Buffer (2X) 25 μl Adaptors (15 M)* 10 μl Quick T4 DNA Ligase 5 μl Total volume 50 μl - Incubate in a thermal cycler for 15 minutes at 20°C.
- Vortex AMPure XP beads to resuspend.
- Add 90 μl (1.8X) of resuspended AMPure XP beads to the ligation reaction (~53 μl). Mix thoroughly on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of 80% freshly prepared ethanol to the tube/pcr plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target by adding 105 μl sterile water to the beads for bead-based size selection as detailed In the next section, or at desired volume for size selection using E-Gel size select gels or standard 2% agarose gels. Mix well on a vortex mixer or by pipetting up and down, and put the tube/pcr plate in the magnetic stand until the solution is clear.
- Transfer 100 μl of supernatant (or desired volume) to a new tube/well, and
proceed to bead based size selection.
Alternatively, adaptor ligated DNA can be purified on one purification column. Elute in sterile water in a volume desired for subsequent size selection.
tttttSize Select Adaptor Ligated DNA Using AMPure XP Beads
| Insert Size |
150 bp |
200 bp |
250 bp |
300 bp |
400 bp |
500 bp |
700 bp |
| Total library size (insert + adaptor) |
270 bp |
320 bp |
370 bp | 420 bp | 530 bp |
660 bp |
820 bp |
| Bead: DNA ratio* 1st bead selection |
0.9X | 0.8X | 0.7X | 0.6X | 0.55X | 0.5X |
0.45X |
| Bead: DNA ratio* 2nd bead selection |
0.2X | 0.2X | 0.2X | 0.2X |
0.15X | 0.15X | 0.15X |
Caution: the following size selection protocol is for libraries with 200 bp inserts only. For libraries with larger fragment inserts, please optimize bead: DNA ratio accordingly.
- Add 80 μl (0.8X) resuspended AMPure XP beads to 100 μl DNA solution. Mix well on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Place the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully transfer the supernatant to a new tube/well (Caution: do not discard the supernatant). Discard beads that contain the large fragments.
- Add 20 μl (0.2X) resuspended AMPure XP beads to the supernatant, mix well and incubate for 5 minutes at room temperature.
- Put the tube/PCR plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets (Caution: do not discard beads).
- Add 200 μl of freshly prepared 80% ethanol to the tube/PCR plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 6 once.
- Air dry beads for 10 minutes while the tube/PCR plate Is on the magnetic stand with the lid open.
- Elute DNA target from the beads into 25 μl sterile water or 0.1X TE Buffer.
Mix well on a vortex mixer or by pipetting up and down, and put the tube/PCR
plate in the magnetic stand until the solution is clear.
Note: Be sure not to transfer any beads. Trace amounts of bead carry-over may affect the optimal performance of the polymerase used in the NEBNext High-Fidelity 2X PCR Master Mix in the subsequent PCR step. - Transfer 20 μl of the supernatant to a clean PCR tube and proceed to
enrichment.
Alternatively, adaptor ligated DNA can be size selected using a number of other methods including E-Gel size select gels or standard 2% agarose gels. Purify DNA sample on one column and elute in 22 μl of sterile water or elution buffer.
- Mix the following components in a sterile microfuge tube:
*NEBNext High-Fidelity 2X PCR Master Mix will be replacing Phusion High-Fidelity PCR Master Mix. Both vials will be supplied for a limited time.DNA 20 μl Primer 1 (25 μM) 2.5 μl Primer 2 (25 μM) 2.5 μl NEBNext High-Fidelity 2X PCR Master Mix* 25 μl Total volume 50 μl
- PCR cycling conditions
*If library construction was performed with 5 μg of starting material, use 4 cycles of amplification. If starting material was 1 μg, use 6-8 cycles of amplification. However, optimization of PCR cycle number may be required to avoid over-amplification.Cycle step Temp. Time Cycles Initial denaturation 98°C 30 sec 1 Denaturation
Annealing
Extension98°C
65°C
72°C10 sec
30 sec
30 sec4–8* Final extension
72°C
4°C5 min
hold1
- Vortex AMPure XP beads to resuspend.
- Add 50 μl (1X) of resuspended AMPure XP beads to the PCR reactions (~50 μl). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/ PCR plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of freshly prepared 80% ethanol to the tube/PCR plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry the beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target from the beads into 30 μl of 0.1X TE. Mix well on a vortex mixer or by pipetting up and down, and put the tube/PCR plate in the magnetic stand until the solution is clear.
- Transfer 25μl of the supernatant to a clean LoBind tube, and store at
-20°C.
Alternatively, adaptor ligated DNA can be purified on one purification column. Purify DNA on one QIAquick column and elute in 27 μl of 0.1X TE Buffer. - Dilute the library 20 fold with nuclease free water, and assess the library
quality on a Bioanalyzer (high sensitivity chip). Check that the
electropherogram shows a narrow distribution with a peak size approximately
300–320 bp.
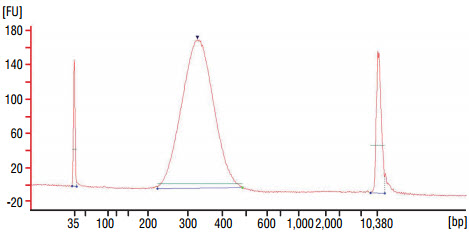
Figure 1: Example of DNA library size distribution on a Bioanalyzer.

