Overcoming the challenges of applying target enrichment for translational research
Target enrichment is used to describe a variety of strategies to selectively isolate specific genomic regions of interest for sequencing analysis. The wide array of approaches presents challenges in selecting the appropriate technology for the growing number of research and clinical applications to which the sequencing data will ultimately be applied.
Introduction
In recent years, several techniques have emerged to enrich for specific genes of interest. When determining the appropriate target enrichment technology to use, one must first consider the primary goal of the study. For example, different approaches will be used if the aim is to identify known variants already shown to have clinical implications, versus discovering novel nucleic acid variants that may be associated with a given phenotype. Variant identification lends itself to more focused enrichment strategies, while variant discovery is driven by trade-offs between sequencing costs and target territory, as well as available sample cohort sizes for a given study.
As translational research seeks to bridge funda-mental laboratory research and clinical treatment regimens for patients, there is an emerging need to balance discovery of novel nucleic acid variants, identification of known variants, and studies aimed at revealing associations with clin-ical phenotypes. Recent advances in sequencing technologies have revolutionized the field of ge-nomic research, making tractable the application of whole genome and whole exome sequenc-ing for broad discovery of germline genomic variants. However, despite these advances, the oncology field is fraught with the complexity of detangling the underpinnings of tumorigenesis, progression, and resistance mechanisms driven by somatic variants present at extremely low abundance in mixtures of malignant and stromal cells. These complexities necessitate increases in the depth of sequencing coverage to confident-ly call somatic variants, making broader scale approaches infeasible from an economic and practical standpoint.
To overcome these challenges, focused gene pan-els are being applied to patient samples. The size of the panel is highly variable, trending toward decreased genomic content as assays progress from pure research and discovery applications to clinical diagnostic assays. Furthermore, clinical applications raise the question of incidental findings and how to report them, introducing challenges for diagnostic assays based on sequencing entire genomes. This trend demonstrates the practical need for continued use of target enrichment strategies across the gamut of translational research activities.
Target Enrichment Approaches
There are a number of different target enrich-ment approaches that can be grouped into three generalized categories: in-solution hybridization, multiplex PCR, and “alternative approaches”, which span a wide variety of techniques.
In-solution hybridization-based approaches, originally developed for whole exome sequenc-ing, use biotinylated oligonucleotides to capture genomic regions of interest (1). Commercial-ly-available kits use DNA or RNA baits ranging from 50-150 nucleotides. Researchers have adapted this technique for more focused panels, ranging down to tens of kilobases in target territory, with limited success in maintaining specificity for target regions.
Multiplex PCR-based enrichment is most often employed for highly focused panels targeting a smaller territory than in-solution hybridization, and is typically limited to 150-200 amplicons (2). Using a pool of primers, enrichment is accomplished through PCR amplification of the targeted regions, which is followed by adaptor ligation or a second round of PCR using tailed primers to include sequencing adaptors. Scaling this technology has presented a challenge in maintaining target coverage uniformity.
A number of alternative approaches have been developed in an attempt to bridge the gap be-tween hybridization and PCR-based approaches.
Examples of these hybrid approaches include multiplex extension ligation (3), molecular inversion probes (MIPS)/padlock probes (4), nested patch PCR (5), and selector probes (6). These technologies can be broadly characterized as having more complex workflows, requiring splitting of samples into separate reactions, and creating challenges in target coverage uniformity.
NEBNext Direct® for target enrichment
NEBNext Direct is a novel target enrichment method that addresses several drawbacks that exist in alternative enrichment technologies (Table 1). Enrichment is achieved through direct hybridization of biotinylated DNA baits to denatured, fragmented molecules, which are subsequently captured using magnetic streptavidin beads (Figure 1, page 3). Unlike alternative in-solution hybridization protocols, the NEBNext Direct protocol does not require library preparation prior to hybridization of oligonucleotide probes. This feature reduces the overall amount of amplification that is required throughout the protocol and enables single-stranded DNA to be captured along with denatured, double-stranded DNA.
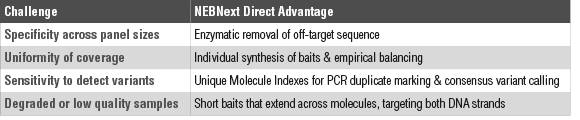
Conversion of captured fragments to sequence-ready libraries is achieved by the ligation of a loop adaptor to the proximal 3´ end of the captured molecule. During these steps, the bait/target molecules remain bound to the magnetic streptavidin beads and are processed in a single reaction tube. This eliminates sample loss and improves overall conversion efficiency.
Following ligation of the 3´ adaptor, the bait is extended across the entirety of the captured molecule, resulting in double stranded DNA that is ready for ligation of the 5´ unique molecular identifier (UMI) adaptor. This adaptor contains a 12 bp random sequence that is incorporated discretely into each molecule, indexing each molecule prior to amplification. This index can be used to identify duplicate molecules, thereby reducing artifacts that can lead to false positive variant calls.
Once the 5´ adaptor is ligated, the 3´ loop adaptor is cleaved, and the target molecule is PCR amplified off of the bait complex. It is important to note that the bait strand is not perpetuated through the PCR amplification and is not present in the final, sequencer-ready library.
The coverage plots of NEBNext Direct libraries are unique for a hybridization-based approach in that reads have a defined 3´ end, resulting in coverage plots that resemble PCR-based libraries, yet the approach allows for flexibility in tiling across longer targets. Disambiguation of PCR duplicates is accomplished by two features of the NEBNext Direct library: A variable 5´ end and a 12 bp randomized UMI that is incorporated into the 5´ adaptor.
Challenges of Target Enrichment for Translational Research
Specificity of target enrichment
For any study that necessitates enrichment of spe-cific targets over more comprehensive sequencing approaches, specificity becomes more important as it directly translates to the amount of sequencing required to achieve the minimum coverage thresh-old to reliably detect variants of a given frequen-cy. Specificity is typically measured by looking at the percentage of sequencing data that is derived from the targeted regions relative to the data that is aligned to other parts of the reference genome.
Enrichment of genomic regions is typically achieved by either amplifying the desired regions through PCR to generate enough copies of the targeted regions over the untargeted regions, or through hybridization of complementary biotinylated oligonucleotide probes to fragmented DNA molecules, where specificity is driven through careful control of melting temperatures and buffer composition to promote hybridization.
Specificity for target regions is enhanced using NEBNext Direct through both the hybridization of specific baits, as well as through enzymatic removal of off-target sequence. The enzymatic treatment removes both off-target sequence of molecules unbound to baits, as well as the regions of molecules upstream of where the baits are bound. This additional means of driving speci-ficity enables the bait hybridization to be shorter, lasting only 90 minutes in duration. This differs from a typical hybridization-based approach, in which randomly fragmented molecules are captured overnight, and without any removal of upstream off-target sequences, read coverage resembles a normal distribution.
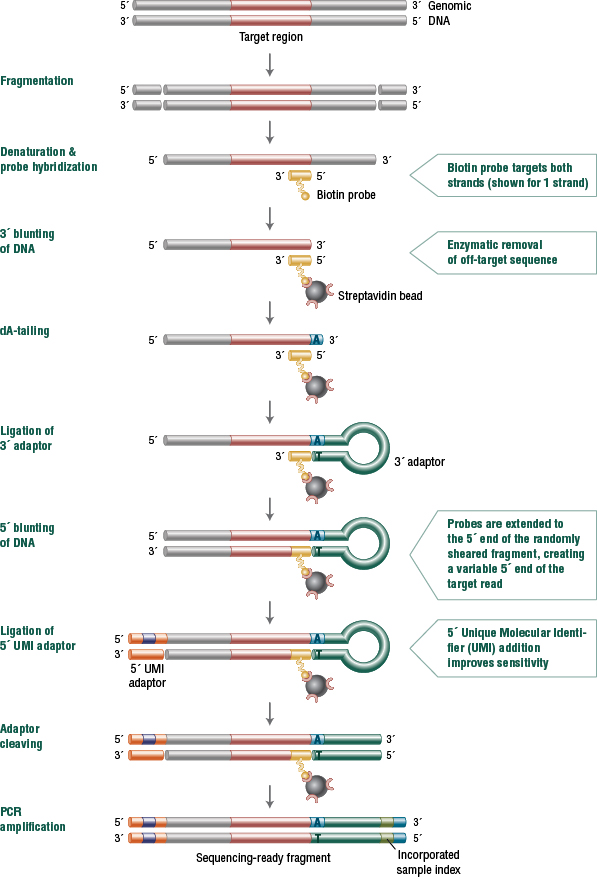
While specificity for targeted regions using tradi-tional hybridization approaches is typically quite high for larger panels up to whole exome, speci-ficity typically decreases as the size of the target-ed region decreases. Thus, smaller panels typically result in an increased proportion of sequencing lost to off-target regions. In contrast, the NEB-Next Direct approach maintains high specificity across a broad range of target territory, from single genes or exons to hundreds of kilobases, eliminating the need to use different technologies for different panels (Table 2).
Table 2: Specificity and uniformity of NEBNext Direct panels
Uniformity of coverage across targets
One of the drawbacks to many available target enrichment methods is the inability to enrich different targets with equivalent efficiency. The result requires an increase in the overall coverage for all targets to achieve the minimum depth of coverage required to reliably call variants. One of the main factors influencing coverage uneven-ness is the sequence composition of the targeted regions themselves, with different efficiencies for sequences comprised of GC or AT rich regions.
Depending on the approach, the target enrich-ment strategy being employed may be more or less susceptible to the need for balancing melting temperatures across any complementary oligonucleotide baits or PCR primers that are employed in the enrichment process. Challenges in uniformity can also arise from any down-stream PCR that is used to generate sufficient material for the sequencing process, as various DNA polymerases demonstrate biases toward targets that may include secondary structure.
Using multiplex PCR-based workflows, primer design is limiting as melting temperatures must match within each panel and primer-primer interactions and primer cross-talk must be consid-ered. These constraints can lead to variations in coverage uniformity between targets. Partitioning individual amplification reactions into emulsion droplets can alleviate some of these constraints and improve target uniformity (12), but this approach requires investment in instrumentation as well as additional workflow steps.
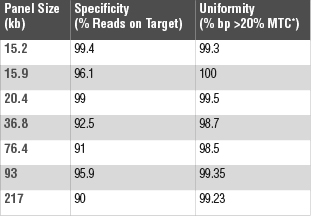
Oligonucleotides utilized during NEBNext Direct enrichment are individually synthe-sized, which enables bait pools to be carefully optimized based on empirical testing. Individual baits are balanced, allowing fine tuning of target coverage. Additionally, the bait design algorithm optimizes new bait design based on outcomes from prior results. Further, because the specificity is not solely driven through melting tempera-tures alone, NEBNext Direct allows increased flexibility in bait design.
The result is coverage across targets that can be optimized, demonstrating high degrees of uniformity and diminishing the overall amount of sequencing required to identify nucleic acid variants (Figure 2).
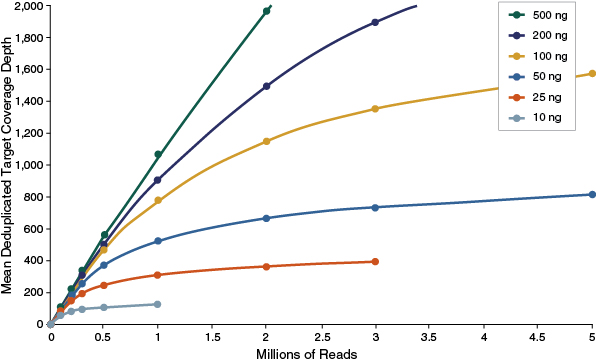
Perhaps the most critical aspect is the sensitivity of an approach to detect nucleic acid variants, as this is often the primary goal of studies in humans where target enrichment is employed. This is measured as the ability of an assay to detect nucleic acid variants that are present at a given frequency, referred to as variant allele frequency (VAF) or mutation allele frequency (MAF). Biologically, in the context of solid tumors, this is a function of the mixture of stromal and tumor cells, as well as the heterogeneity of tumor cells, and the existence of subclonal variants that are associated with tumorigenesis. Utilization of sequence data for the approximation of allele frequency is achieved through counting of sequence reads that possess a given variant. Quantitative assessment of sequence reads is challenged through the presence of duplicate molecules, or molecules that are identified through sequencing as having the same genomic coordinates. Depending on the target enrichment method that was employed to prepare the samples for sequencing, disambiguation of molecules that have arisen from discrete copies of genomic DNA versus those resulting from PCR amplification can be difficult or impossible to ascertain.
Disambiguation of PCR duplicates is ac-complished by two features of the NEBNext Direct library: A variable 5´ end and a 12 bp randomized UMI that is incorporated into the 5´ adaptor. The amount of coverage one can expect from a given panel should be measured once duplicate molecules are removed in order to determine if the coverage is deep enough to reliably call a variant as a true-positive variant (Figure 3).
Difficult sample types
Whether for research or clinical applications, translational genomics often examines samples that are derived from patients. Patient tissue can be compromised by processes used to collect, preserve, store, extract nucleic acids from, and ultimately prepare for sequencing-based assays.
a broad range of inputs.
Conclusion
NEBNext Direct target enrichment overcomes several challenges translational researchers face in selectively enriching for certain genomic targets for clinical research. Providing the flexibility to use a single approach across a wide range of target content, NEBNext Direct allows enrichment of a single gene, up to panels comprised of hundreds of genes, without compromising performance as targets change. NEBNext Direct provides the specificity and coverage uniformity to maximize sequencing efficiency, in order to realize the benefits of target enrichment. Furthermore, intrinsic properties of the approach lend themselves to improved sensitivity, and have proven amenable to challenging sample types, typical of translational workflows. Combining the best aspects of hybridization-based enrichment and multiplex PCR enrichment, without the trade-offs, NEBNext Direct is a single-day, easy-to-use protocol that can be applied to advance translational research.
View a PDF of this white paper.
References
- Gnirke, A., et al. (2009) Nature Biotechnology 27, 182–189.
- Mertes, F., et al. (2011) Briefings in Functional Genomics 10, 374–386.
- Shen, M.-J. R., et al. (2011) Multiplex nucleic acid reactions. US Patent 7955794 B2.
- Porreca, G. J., et al. (2007) Nature Methods 4, 931–936.
- Varley, K. E., and Mitra, R. D. (2008) Genome Research 18, 1844–1850.
- Johansson, H., et al. (2011) Nucleic Acids Research 39:e8.
- Srinivasan, M., et al. (2002) The American Journal of Pathology 161, 1961–1971.
- Costello, M, et al. (2013) Nucleic Acids Research 41(6):e67.
- Chen, L., et al. (2017) Science 355, 752-756.
- Stiller, M., et al. (2016) Oncotarget 37(7), 59115-59128.
- Burnham, P., et al. (2016) Scientific Reports 6, 27859.
- Tewhey, R., et al. (2009) Nature Biotechnology 27, 1025–1031.
