Minding your caps and tails
Applications of synthetic mRNA have grown and become considerably diversified in recent years. Examples include the generation of pluripotent stem cells (1-3), vaccines and therapeutics (4-5), and CRISPR/Cas9 genome editing applications (6-8). The basic requirements for a functional mRNA – a 7-methylguanylate cap at the 5´ end and a poly(A) tail at the 3´ end – must be added in order to obtain efficient translation in eukaryotic cells. Additional considerations can include the incorporation of internal modified bases, modified cap structures and polyadenylation strategies. Strategies for in vitro synthesis of mRNA vary according to the desired scale of synthesis. This article discusses options for the selection of reagents and the extent to which they influence synthesized mRNA functionality.
by Breton Hornblower, Ph.D., G. Brett Robb, Ph.D. and George Tzertzinis, Ph.D., New England Biolabs, Inc
A nascent mRNA, synthesized in the nucleus, undergoes different modifications before it can be translated into proteins in the cytoplasm. For a mRNA to be functional, it requires modified 5´ and 3´ ends and a coding region (i.e., an open reading frame (ORF) encoding for the protein of interest) flanked by the untranslated regions (UTRs). The nascent mRNA (pre-mRNA) undergoes two significant modifications in addition to splicing. During synthesis, a 7-methylguanylate structure, also known as a “cap”, is added to the 5´ end of the pre-mRNA, via 5´ → 5´ triphosphate linkage. This cap protects the mature mRNA from degradation, and also serves a role in nuclear export and efficient translation.
The second modification occurs posttranscriptionally at the 3´ end of the nascent RNA molecule, and is characterized by addition of approximately 200 adenylate nucleotides (poly(A) tail). The addition of the the poly(A) tail confers stability to the mRNA, aids in the export of the mRNA to the cytosol, and is involved in the formation of a translation-competent ribonucleoprotein (RNP), together with the 5´ cap structure. The mature mRNA forms a circular structure (closed-loop) by bridging the cap to the poly(A) tail via the cap-binding protein eIF4E (eukaryotic initiation factor 4E) and the poly(A)- binding protein, both of which interact with eIF4G (eukaryotic initiation factor 4G), (Figure 1, (9)).
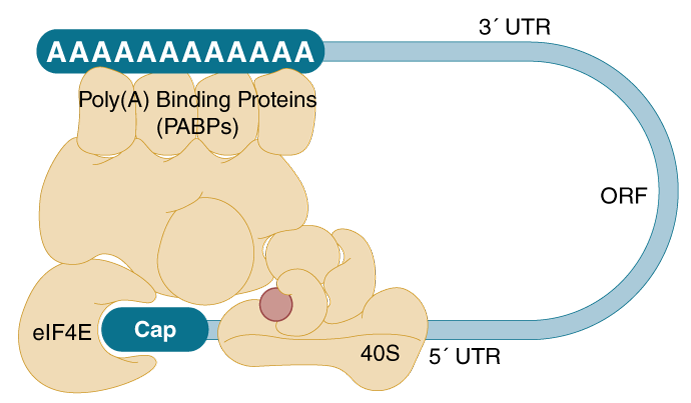
RNA can be efficiently synthesized in vitro (by in vitro transcription, IVT) with prokaryotic phage polymerases, such as T7, T3 and SP6. The cap and poly(A) tail structures characteristic of mature mRNA can be added during or after the synthesis by enzymatic reactions with capping enzymes and Poly(A) Polymerase (NEB #M0276), respectively.
There are several factors to consider when planning for IVT-mRNA synthesis that will influence the ease-of-experimental setup and yield of the final mRNA product. These are discussed in the following sections.
DNA template
The DNA template provides the sequence to be transcribed downstream of an RNA polymerase promoter. There are two strategies for generating transcription templates: PCR amplification and linearization of plasmid with a restriction enzyme (Figure 2). Which one to choose will depend on the downstream application. In general, if multiple sequences are to be made and transcribed in parallel, PCR amplification is recommended as it generates many templates quickly. On the other hand, if large amounts of one or a few templates are required, plasmid DNA is recommended, because of the relative ease of producing large quantities of high quality, fully characterized plasmids. There are different versions of plasmids available that allow for propagation of homopolymeric A-tails of defined length (1).
PCR allows conversion of any DNA fragment to a transcription template by appending the T7 (or SP6) promoter to the forward primer (Figure 2A). Additionally, poly(d)T-tailed reverse primers can be used in PCR to generate transcription templates with A-tails. This obviates the need for a separate polyadenylation step following transcription. Repeated amplifications should, however, be avoided to prevent PCR-generated point mutations. Amplification using PCR enzymes with the highest possible fidelity, such as Q5® High-Fidelity DNA Polymerase (NEB #M0491), reduces the likelihood of introducing such mutations (2).
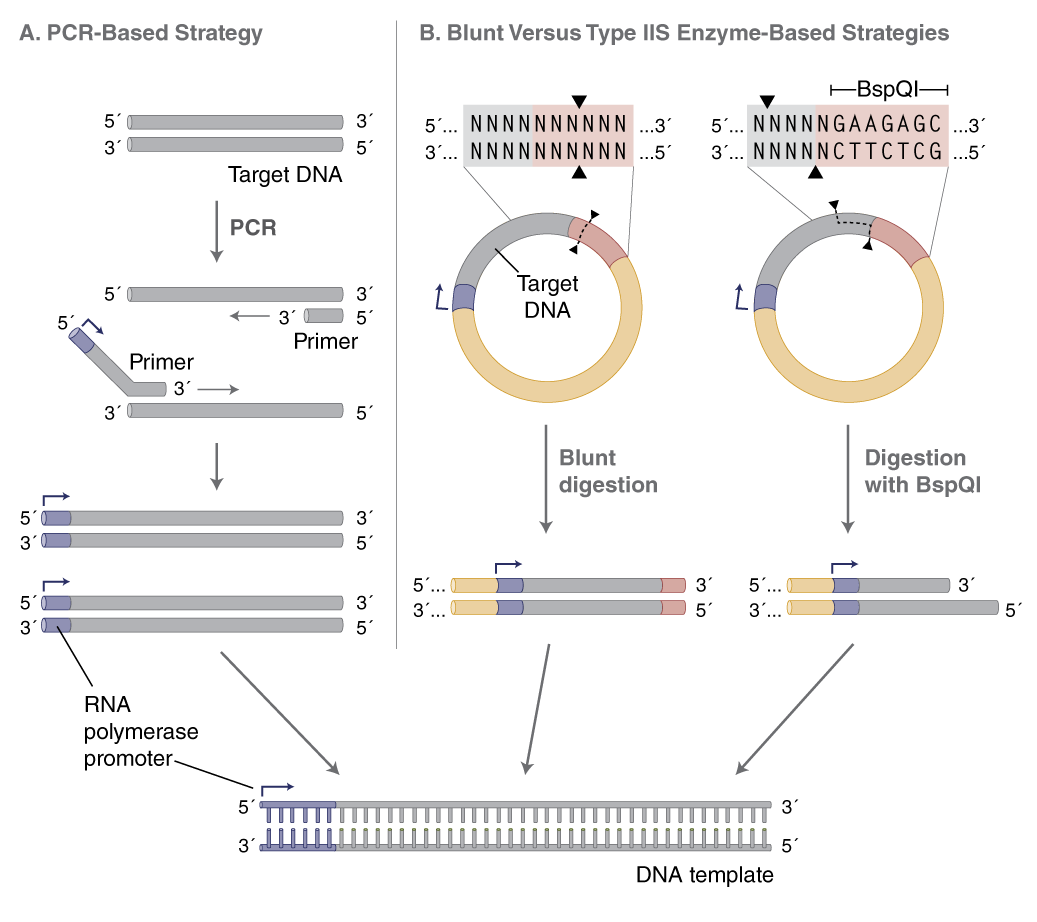
(B) When using plasmid DNA as a template, linearize with an enzyme that produces blunt or 5´-overhanging ends. Using a type IIS restriction enzyme (e.g., BspQI) allows RNA synthesis with no additional 3´-nucleotide sequence from the restriction site.
The quality of the PCR reaction can be assessed by running a small amount on an agarose gel, and DNA should be purified before in vitro transcription using a spin column or magnetic beads (e.g., AMPure® beads). Multiple PCR reactions can be purified and combined to generate a DNA stock solution that can be stored at -20°C and used as needed for in vitro transcription.
Plasmid templates are convenient if the template sequence already exists in a eukaryotic expression vector also containing the T7 promoter (e.g., pcDNA vector series). These templates include 5´- and 3´-untranslated regions (UTR), which are important for the expression characteristics of the mRNA.
Plasmid DNA should be purified and linearized downstream of the desired sequence, preferably with a restriction enzyme that leaves blunt or 5´ overhangs at the 3´ end of the template. These are favorable for proper run-off transcription by T7 RNA Polymerase (NEB #M0274), while 3´ overhangs may result in unwanted transcription products. To avoid adding extra nucleotides from the restriction site to the RNA sequence, a Type IIS restriction enzyme can be used (e.g., BspQI, NEB #R0712), which positions the recognition sequence outside of the transcribed sequence (Figure 2B, page 2). The plasmid DNA should be completely digested with the restriction enzyme, followed by purification using a spin column (e.g., Monarch® PCR & DNA Cleanup Kit (5 μg) NEB #T1030) or phenol extraction/ethanol precipitation. Although linearization of plasmid involves multiple steps, the process is easier to scale for the generation of large amounts of template for multiple transcription reactions.
In vitro transcription
There are two options for the in vitro transcription (IVT) reaction depending on the capping strategy chosen: standard synthesis with enzyme-based capping following the transcription reaction (post-transcriptional capping) or incorporation of a cap analog during transcription (co-transcriptional capping) (Figure 3). Method selection will depend on the scale of mRNA synthesis required and number of templates to be transcribed.
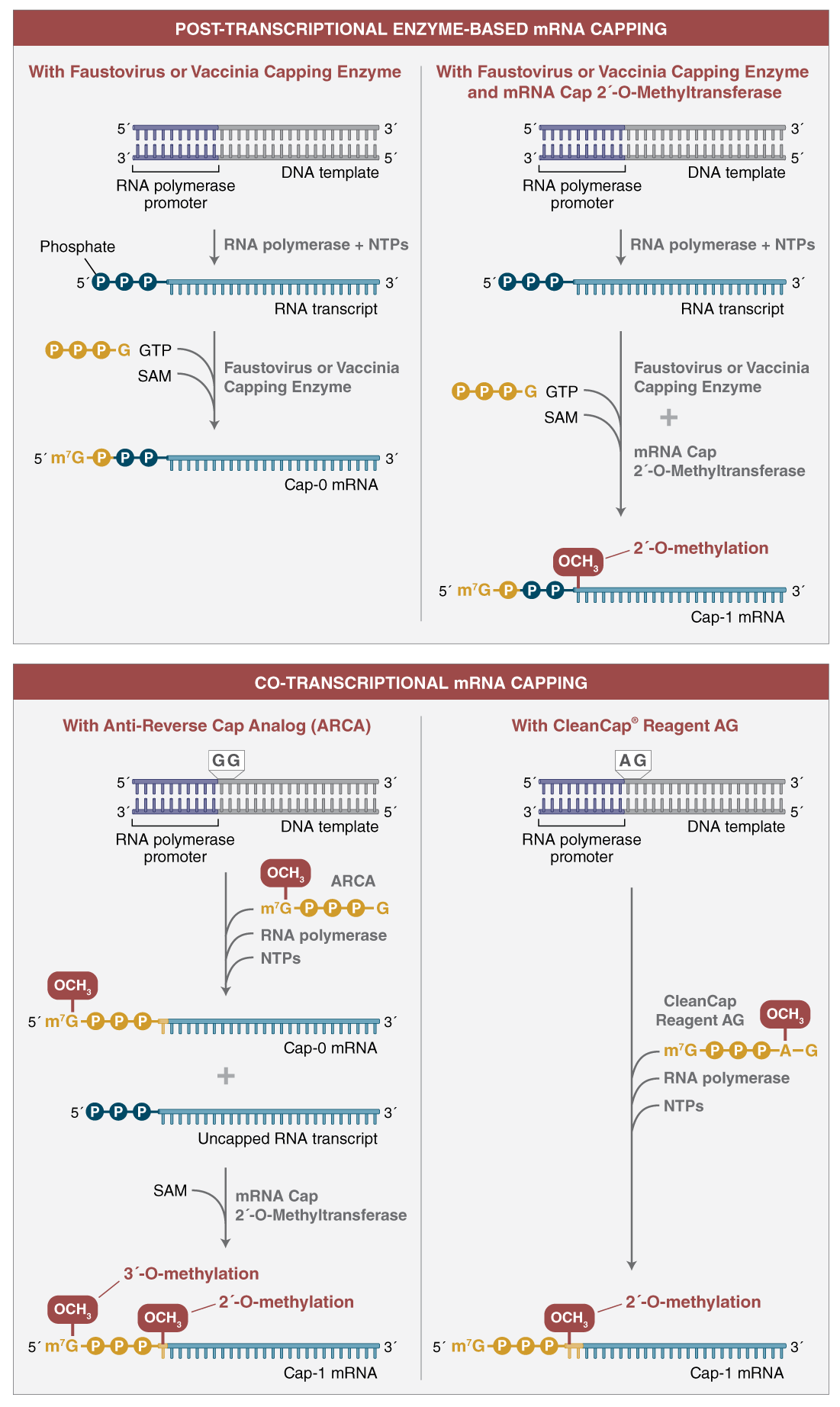
Co-transcriptional capping (bottom) uses an mRNA cap analog, shown in yellow, in the transcription reaction. For ARCA (anti-reverse cap analog) (left),the cap analog is incorporated as the first nucleotide of the transcript. ARCA contains an additional 3´-O-methyl group on the 7-methylguanosine to ensure incorporation in the correct orientation. The 3´-O-methyl modification does not occur in natural mRNA caps. Compared to reactions not containing cap analog, transcription yields are lower. ARCA- capped mRNA can be converted to cap 1 mRNA using mRNA cap 2´-O-MTase and SAM in a subsequent reaction. CleanCap Reagent AG (right) uses a trinucleotide cap analog that requires a modified template initiation sequence. A natural Cap-1 structure is accomplished in a co-transcriptional reaction.
Transcription for enzyme-based capping (post-transcriptional capping)
Standard RNA synthesis reactions produce the highest yield of RNA transcript (typically ≥100 μg per 20 μl in a 1 hr reaction using the HiScribe®; Quick T7 High Yield RNA Synthesis Kit, NEB #E2050S). Transcription reactions are highly scalable, and can be performed using an all-inclusive kit (e.g., HiScribe kits), or individual reagents. More information on the HiScribe kits can be found later in the article.
Following transcription, the RNA is treated with DNase I (NEB #M0303) or DNase I-XT (NEB #M0570) to remove the DNA template, and purified using an appropriate column, kit or magnetic beads, prior to capping. This method produces high yields of RNA with 5´-triphosphate termini that must be converted to cap structures. In the absence of template-encoded poly(A) tails, transcripts produced using this method bear 3´ termini that also must be polyadenylated in a separate enzymatic step, as described below in “A-tailing using E. coli Poly(A) Polymerase”.
Transcription with dinucleotide co-transcriptional capping
In co-transcriptional capping, a cap analog is introduced into the transcription reaction, along with the four standard nucleotide triphosphates, in an optimized ratio of cap analog to GTP 4:1. This allows initiation of the transcript with the cap structure in a large proportion of the synthesized RNA molecules. This approach produces a mixture of transcripts, of which ~80% are capped, and the remainder have 5´-triphosphate ends. Decreased overall yield of RNA products results from the lower concentration of GTP in the reaction (Figure 4).
There are several cap analogs used in co-transcriptional RNA capping (3,4). The most common are the standard 7-methyl guanosine (m7G) cap analog and anti-reverse cap analog (ARCA), also known as 3´ O-me 7-meGpppG cap analog. ARCA is methylated at the 3´ position of the m7G, preventing RNA elongation by phosphodiester bond formation at this position.
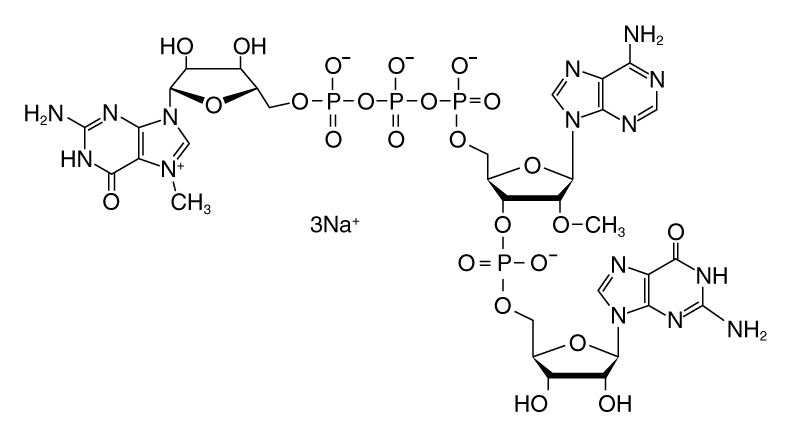
Thus, transcripts synthesized using ARCA contain 5´-m7G cap structures in the correct orientation, with the 7-methylated G as the terminal residue. In contrast, the m7G cap analog can be incorporated in either the correct or the reverse orientation.
HiScribe T7 ARCA mRNA Synthesis kits (NEB #E2060 and #E2065) contain reagents, including an optimized mix of ARCA and NTPs, for streamlined reaction setup for synthesis of co-transcriptionally capped RNAs.
Transcription with CleanCap® reagent AG co-transcriptional capping
The use of CleanCap reagent AG results in significant advantages over traditional dinucleotide co-transcriptional capping. CleanCap Reagent AG is a trinucleotide with a 5´-m7G joined by a 5´-5´ triphosphate linkage to an AG sequence. The adenine has a methyl group on the 2´-O position (Figure 4). The incorporation of this trinucleotide in the beginning of a transcript results in a Cap-1 structure.
In order to use CleanCap Reagent AG in an in vitro transcription reaction the template must contain an AG in place of a GG following the T7 promoter in the initiation sequence.
Unlike traditional co-transcriptional capping, reduction of GTP concentration is not required and therefore yield is higher and high capping efficiencies, >95%, are achieved (Figure 5).
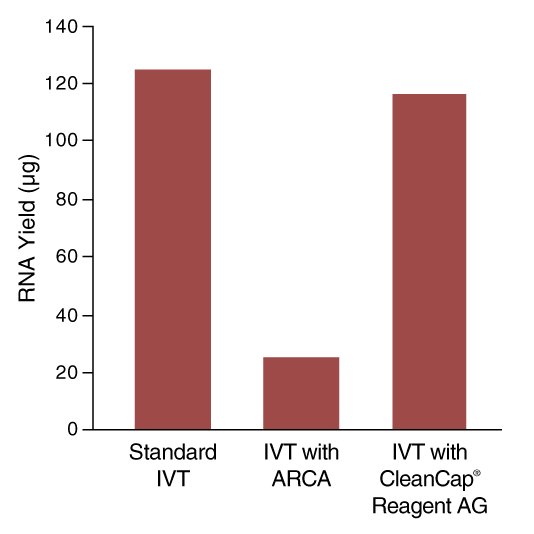
Transcription with complete substitution with modified nucleotides
RNA synthesis can be carried out with a mixture of modified nucleotides in place of the regular mixture of A, G, C and U triphosphates. For expression applications, the modified nucleotides of choice are the naturally occurring 5´-methylcytidine and/or pseudouridine in the place of C and U, respectively. These have been demonstrated to confer desirable properties to the mRNA, such as increased mRNA stability, increased translation, and reduced immune response in the key applications of protein replacement and stem-cell differentiation (1). It is important to note that nucleotide choice can influence the overall yield of mRNA synthesis reactions.
Fully substituted RNA synthesis can be achieved using the HiScribe T7 mRNA Kit with CleanCap Reagent AG (NEB #E2080), HiScribe T7 High-Yield RNA Synthesis Kit (NEB #E2040) or HiScribe SP6 RNA Synthesis Kit (NEB #E2070) in conjunction with NTPs with the desired modification. Transcripts made with complete replacement of one or more nucleotides may be post-transcriptionally capped (see next section), or may be co-transcriptionally capped by including CleanCap Reagent AG, ARCA or another cap analog, as described previously.
If partial replacement of nucleotides is desired, the HiScribe T7 ARCA mRNA Synthesis Kits (NEB #E2060 and #E2065), may be used with added modified NTPs, to produce co-transcriptionally capped mRNAs, as described above. Alternatively, the HiScribe T7 Quick RNA Synthesis Kit (NEB #E2050) may be used to prepare transcripts for post-transcriptional capping.
Post-transcriptional capping and Cap-1 methylation
Post-transcriptional capping is often performed using the mRNA capping enzyme from Vaccinia virus or Faustovirus. These enzymes complex converts the 5´-triphosphate ends of in vitro transcripts to m7G-cap (Cap-0) required for efficient protein translation in eukaryotes. The Faustovirus capping enzyme (NEB #M2081) comprises three enzymatic activities (RNA triphosphatase, guanylyltransferase, guanine N7-methyltransferase) that are necessary for the formation of the complete Cap-0 structure, m7Gppp5´N, using GTP and the methyl donor S-adenosylmethionine. As an added option, the inclusion of the mRNA Cap 2´ O-Methyltransferase (NEB #M0366) in the same reaction results in formation of the Cap-1 structure (m7Gppp5´Nm), a natural modification in many eukaryotic mRNAs responsible for evading cellular innate immune response against foreign RNA. This enzyme-based capping approach results in a high proportion of capped message, and it is easily scalable. The resulting capped RNA can be further modified by poly(A) addition before final purification.
A-tailing using E. coli Poly(A) Polymerase
The poly(A) tail confers stability to the mRNA and enhances translation efficiency. The poly(A) tail can be encoded in the DNA template by using an appropriately tailed PCR primer, or it can be added to the RNA by enzymatic treatment with E. coli Poly(A) Polymerase (NEB #M0276). The length of the added tail can be adjusted by titrating the Poly(A) Polymerase in the reaction (Figure 6).
The importance of the A-tail is demonstrated by transfection of untailed vs. tailed mRNA. When luciferase activity from cells transfected with equimolar amounts of tailed or untailed mRNAs were compared, a significant enhancement of translation efficiency was evident (Figure 6). HiScribe T7 ARCA mRNA Synthesis Kit (with tailing) (NEB #E2060) includes E. coli Poly(A) Polymerase, and enables a streamlined workflow for the enzymatic tailing of co-transcriptionally capped RNA.
For mRNA synthesis from templates with encoded poly(A) tails, the HiScribe T7 ARCA mRNA Synthesis Kit (NEB #E2065) provides an optimized formulation for co-transcriptionally capped transcripts.
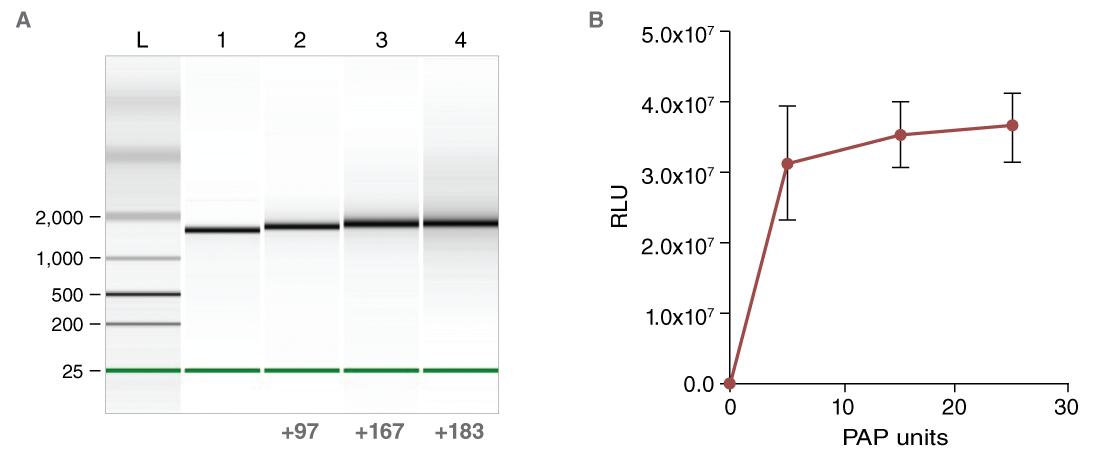
(B) Effect of enzymatic A-tailing on the luciferase reporter activity of CLuc mRNA.
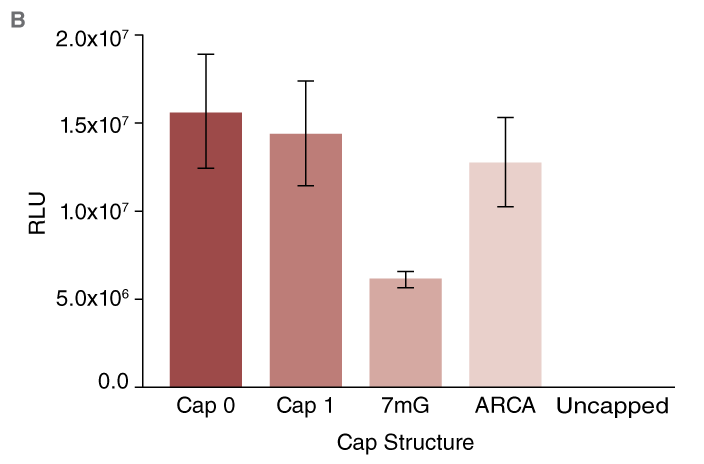
(B) Expression of Cypridina luciferase (CLuc) after capping using different methods. High activity from all capped RNAs is observed.
The effect of capping can be studied by delivering the mRNA to cultured mammalian cells and monitoring its translation. Using RNA encoding secreted luciferases (e.g., Cypridina luciferase, CLuc) the translation can be monitored by assaying its activity in the cell culture medium (Fig. A).
CLuc mRNA was synthesized and capped post-transcriptionally (Cap 0 or Cap 1) or co-transcriptionally (as described above) using standard (7mG) or anti-reverse cap analog (ARCA). For consistency, the mRNAs were prepared from templates encoding poly-A tails of the same length.
After capping, the mRNA was purified using magnetic beads and quantified before transfection into U2OS cells using the TransIT® mRNA transfection reagent following the manufacturer’s protocol. CLuc activity was measured 16 hrs after transfection using the BioLux® Cypridina Luciferase Assay Kit (NEB #E3309).
Virtually no luciferase reporter activity was observed in conditions where uncapped RNA was transfected (Fig. B). In contrast, robust activity was detected from cells transfected with RNA capped using the methods described above. As anticipated, lower activity was observed from cells transfected with mRNA capped using the 7mG cap analog as compared to ARCA-capped mRNA.
Summary
In summary, when choosing the right workflow for your functional mRNA synthesis needs, you must balance your experimental requirements for the mRNA (e.g., internal modified nucleotides) with scalability (i.e., ease-of-reaction setup vs. yield of final product).
In general, co-transcriptional capping of mRNA with template encoded poly(A) tails or post-transcriptional addition of poly(A) tail is recommended for most applications. This approach, using the HiScribe T7 mRNA Kits with CleanCap Reagent AG (NEB #E2080), enables the quick and streamlined production of one or many transcripts with typical yields of ≥90 μg per reaction, totaling ~1.8 mg per kit.
Post-transcriptional mRNA capping with Faustovirus Capping Enzyme or Vaccinia Capping System is well suited to larger scale synthesis of one or a few mRNAs, and is readily scalable to produce gram-scale quantities and beyond. Reagents for in vitro synthesis of mRNA are available in kit form or as separate components to enable research and large-scale production.
Products from NEB are available for each step of the RNA Synthesis Product Workflow. GMP-grade products suitable for manufacture of large scale manufacture of therapeutic mRNA are available through our Customized Solution Group.
View a PDF of this feature article
References:
- Warren, L., et al. (2010) Cell Stem Cell, 7, 618-630.
- Angel, M. and Yanik, M.F. (2010) PLoS One, 5:e11756.
- Yakubov, E., et al. (2010) Biochem. Biophys. Res. Commun. 394, 189.
- Geall, A.J., et al. (2012) Proc. Natl. Acad. Sci. USA, 109, 14604-14609.
- Ramaswamy, S., et al. (2017) Proc. Natl. Acad. Sci. USA, 114, E1941-E1950.
- Ma, Y., et al. (2014) PLoS One, 9:e89413.
- Ota, S., et al. (2014) Genes Cells, 19, 555-564.
- Bassett, A. R., et al. (2013) Cell Rep. 4, 220–228.
- Wells, S.E., et al. (1998) Molecular Cell 2, 135–140.

