Enzymatic Methyl-seq: The Next Generation of Methylome Analysis
by Louise Williams, Ph.D., Yanxia Bei, Ph.D., Heidi E. Church, Nan Dai, Ph.D., Eileen T. Dimalanta, Ph.D., Laurence M. Ettwiller, Ph.D., Thomas C. Evans, Jr., Ph.D., Bradley W. Langhorst, Ph.D., Janine G. Borgaro, Ph.D., Shengxi Guan, Ph.D., Katherine Marks, Julie F. Menin, Nicole M. Nichols, Ph.D., V. K. Chaithanya Ponnaluri, Ph.D., Lana Saleh, Ph.D., Mala Samaranayake, Ph.D., Brittany S. Sexton, Ph.D., Zhiyi Sun, Ph.D., Esta Tamanaha, Ph.D., Romualdas Vaisvila, Ph.D., Erbay Yigit, Ph.D. and Theodore B. Davis, New England Biolabs, Inc.
The identification of cytosine modifications within genomes, especially 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), is important as they are known to have an impact on gene expression. Generally, low levels of methylation near transcription start sites are associated with higher transcription levels, while genes with regulatory regions containing high levels of cytosine modification are expressed at lower levels. The ability to analyze a complete methylome is important for studying diseases, including those associated with cancer, metabolic disorders and autoimmune diseases. Unfortunately, the current technologies for investigating 5mC and 5hmC are sub-optimal and do not permit a thorough evaluation of methylomes.
Bisulfite Sequencing
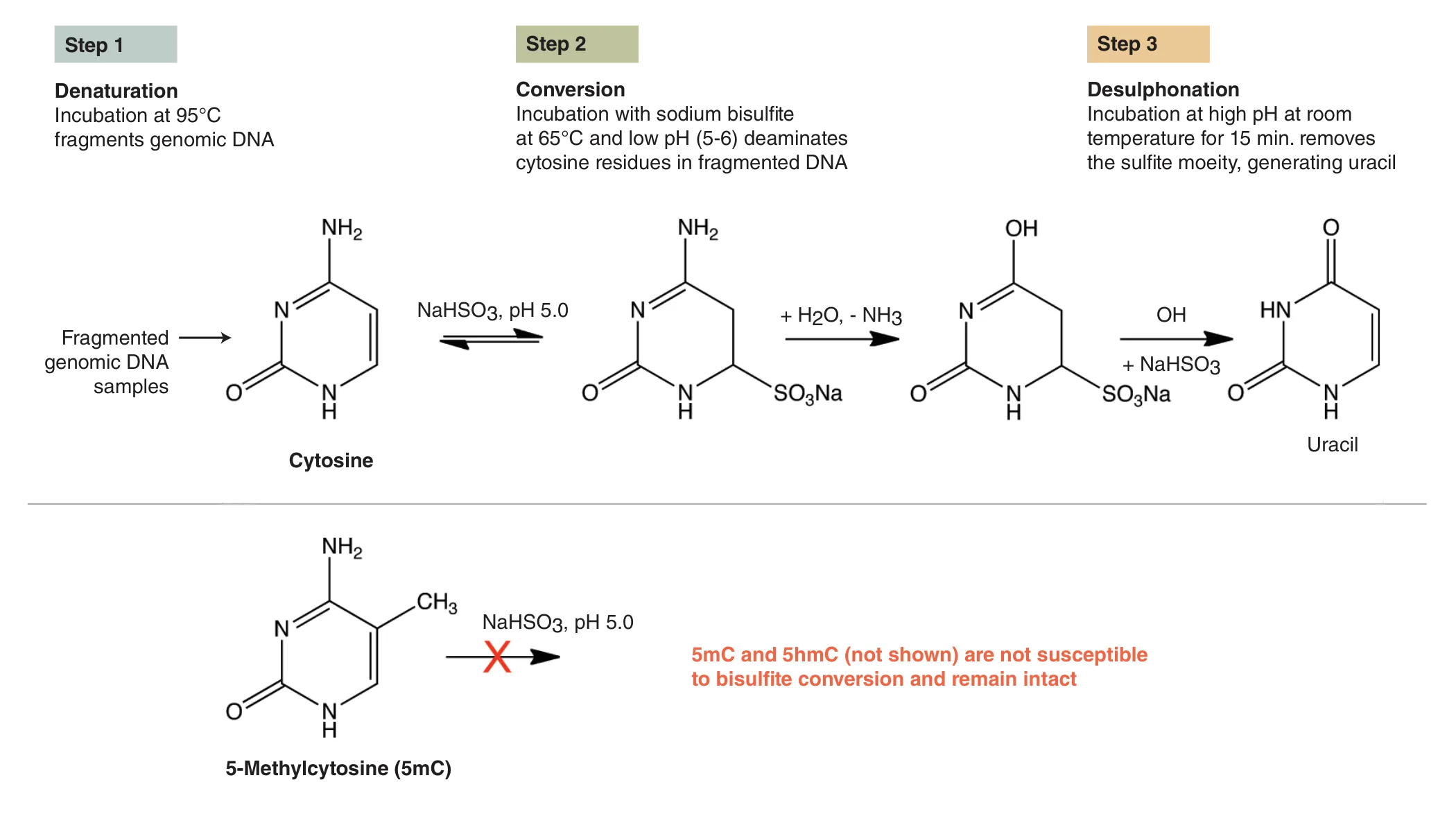
To date, the gold standard in methylome mapping has been bisulfite sequencing. In this method, DNA is chemically treated with sodium bisulfite, which results in the conversion of unmethylated cytosines to uracils, and the resulting uracils are ultimately sequenced as thymines (Figure 1). In contrast, the modified cytosines, 5mC and 5hmC, are resistant to bisulfite conversion, and are sequenced as cytosines (1). While the preparation of bisulfite libraries is relatively straightforward, the libraries have uneven genome coverage and therefore suffer from incomplete representation of cytosine methylation across genomes. This uneven coverage is the result of DNA damage and fragmentation, which is caused by the extreme temperatures and pH during bisulfite conversion. Sequenced bisulfite libraries typically have skewed GC bias plots, with a general under-representation of G- and C-containing dinucleotides and over-representation of AA-, AT- and TA-containing dinucleotides, as compared to a non-converted genome (2). Therefore, the damaged libraries do not adequately cover the genome, and can include many gaps with little or no coverage. Increasing the sequencing depth of these libraries can recover some missing information, but at steep sequencing costs. These bisulfite library limitations have driven the development of new approaches for studying methylomes.
Alternative Methods For Detecting 5mC AND 5hmC
Additional approaches for investigating methylomes are available that either combine bisulfite conversion with another chemical modification or an enzymatic modification step, or that eliminate bisulfite conversion completely (Table 1).
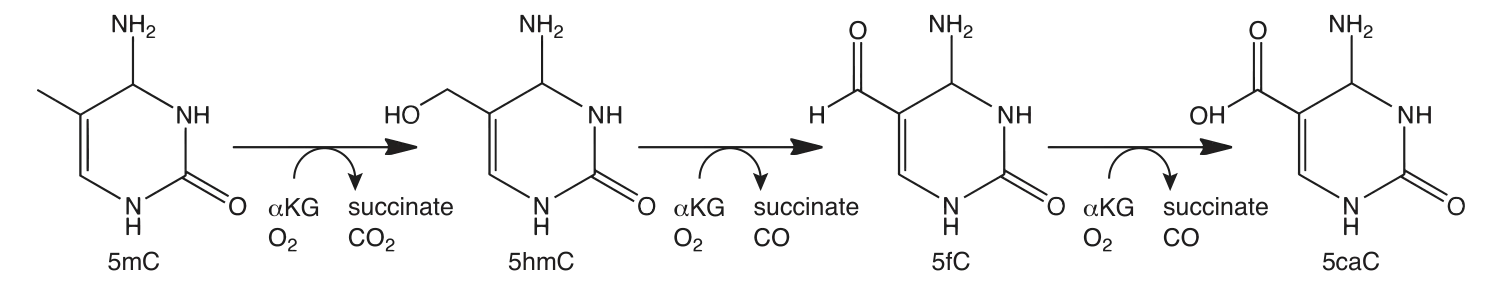
5hmC can be detected using TET-assisted bisulfite sequencing (TAB-seq). Fragmented DNA is enzymatically modified using sequential T4 Phage ß-glucosyltransferase (T4-BGT) and then Ten-eleven translocation (TET) dioxygenase treatments before the addition of sodium bisulfite (3). T4-BGT glucosylates 5hmC to form beta-glucosyl-5-hydroxymethylcytosine (5ghmC) and TET is then used to oxidize 5mC to 5caC (Figure 2). Only 5ghmC is protected from subsequent deamination by sodium bisulfite and this enables 5hmC to be distinguished from 5mC by sequencing.
Oxidative bisulfite sequencing (oxBS) provides another method to distinguish between 5mC and 5hmC (4). The oxidation reagent potassium perruthenate converts 5hmC to 5-formylcytosine (5fC) and subsequent sodium bisulfite treatment deaminates 5fC to uracil. 5mC remains unchanged and can therefore be identified using this method.
Table 1: Summary of alternative methods of methylome analysis
| Sequencing Method | Cytosine Modification | Method of Analysis | Weaknes |
|---|---|---|---|
| TET-assisted bisulfite sequencing (TAB-seq) (3) | 5hmC | Enzymatic treatment with T4-BGT then TET followed by bisulfite treatment | DNA damage and sequencing bias |
| Oxidative bisulfite sequencing (oxBS) (4) | 5hmC | Treatment with an oxidation reagent, followed by bisulfite treatment | DNA damage and sequencing bias |
| APOBEC-coupled epigenetic sequencing (ACE-seq) (5) | 5hmC | Enzymatic treatment with T4-BGT and APOBEC3A | APOBEC3A not commercially available |
| TET-assisted 5-methylcytosine sequencing (TAmC-seq) (6) | 5hmC | Enzymatic treatment, followed by enrichment for 5mC regions | Enriches for 5mC dense regions. Does not currently cover entire genome |
APOBEC-coupled epigenetic sequencing (ACE-seq) excludes bisulfite conversion altogether and relies on enzymatic conversion to detect 5hmC (5). With this method, T4-BGT glucosylates 5hmC to 5ghmC and protects it from deamination by Apolipoprotein B mRNA editing enzyme subunit 3A (APOBEC3A). Cytosine and 5mC are deaminated by APOBEC3A and sequenced as thymine.
Lastly, TET-assisted 5-methylcytosine sequencing (TAmC-seq) enriches for 5mC loci and utilizes two sequential enzymatic reactions followed by an affinity pull-down (6). Fragmented DNA is treated with T4-BGT which protects 5hmC by glucosylation. The enzyme mTET1 is then used to oxidize 5mC to 5hmC, and T4-BGT labels the newly formed 5hmC using a modified glucose moiety (6-N3-glucose). Click chemistry is used to introduce a biotin tag which enables enrichment of 5mC-containing DNA fragments for detection and genome wide profiling.
Libraries made from methods that combine enzymatic and sodium bisulfite identification of cytosine modifications all experience DNA damage and the inherent biases of bisulfite treatment. Furthermore, the described enzymatic methods have additional drawbacks. TAmC-seq is focused on loci and does not discriminate between methylated and unmethylated cytosines in the enriched DNA fragments. ACE-seq probes only 5hmC and requires APOBEC3A for deamination, which is not yet commercially available, making it more difficult to standardize library construction between labs.
Enzymatic Methyl-seq – A New Approach
The enzymatic methyl-seq workflow developed at NEB provides a much-needed alternative to bisulfite sequencing. This method relies on the ability of APOBEC to deaminate cytosines to uracils. Unfortunately, APOBEC also deaminates 5mC and 5hmC, making it impossible to differentiate between cytosine and its modified forms (7,8). In order to detect 5mC and 5hmC, this method also utilizes TET2 and an Oxidation Enhancer, which enzymatically modifies 5mC and 5hmC to forms that are not substrates for APOBEC. The TET2 enzyme converts 5mC to 5caC (Figure 2) and the Oxidation Enhancer converts 5hmC to 5ghmC (9,10,11). Ultimately, cytosines are sequenced as thymines and 5mC and 5hmC are sequenced as cytosines, thereby protecting the integrity of the original 5mC and 5hmC sequence information.
The NEBNext Enzymatic Methyl-seq Kit (EM-seq™) combines NEBNext® Ultra™ II reagents with these two enzymatic steps to construct Illumina® libraries that accurately represent 5mC and 5hmC within the genome. Converted libraries are amplified using NEBNext Q5U DNA polymerase (Figure 3). EM-seq libraries result in a more accurate representation of the methylome, with minimal DNA fragmentation or biases when compared to whole genome bisulfite sequencing (WGBS). The combination of the Ultra II reagents for library prep and the EM-seq conversion allows for lower input amounts compared to most WGBS workflows, with a range of inputs from 10 – 200 ng.

EM-seq Performance
Intact DNA
Several pieces of data suggest that the process of generating EM-seq libraries does not damage DNA in the same way as bisulfite sequencing. EM-seq libraries give higher PCR yields despite using fewer PCR cycles for all DNA input amounts (see page 6), indicating that less DNA is lost during enzymatic treatment and library preparation, as compared to WGBS. Reduced PCR cycles, in turn, translates into more complex libraries and fewer PCR duplicates during sequencing (data not shown). EM-seq libraries also have larger insert sizes than WGBS (Figure 4), which further supports the fact that DNA remains intact.

EM-seq Libraries Have Reduced Bias
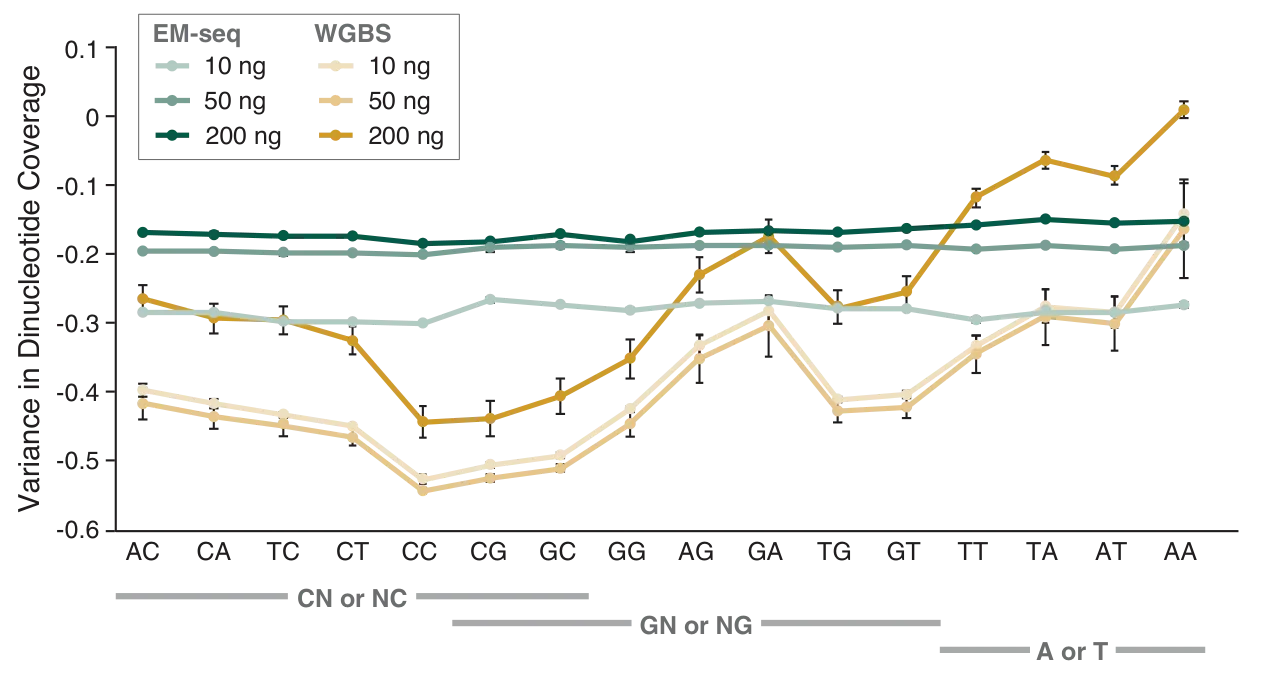
The preservation of DNA integrity is also demonstrated by the GC bias graphs (Figure 5), and the dinucleotide coverage distribution graph (Figure 6). Both of these figures indicate that reduced bias is associated with the EM-seq libraries. The EM-seq libraries have a flat GC bias distribution (Figure 5) with even coverage at both GC and AT rich regions, and do not display a preference for any dinucleotide combination (Figure 6). This is in stark contrast to WGBS, which shows a skewed GC bias profile along with the previously mentioned dinucleotide biases. Reduced library bias improves the mapping and therefore coverage of CpGs.

CpG Detection
Human DNA is methylated almost exclusively in CpG contexts. EM-seq global CpG methylation levels for human NA12878 DNA are consistent with WGBS libraries (Figure 7A), indicating that EM-seq libraries accurately detect methylation. The more striking difference between EM-seq and WGBS libraries becomes apparent when the focus is shifted to CpG coverage. EM-seq libraries detect more CpGs to a higher depth of coverage than WGBS libraries (Figure 7B). The ability to detect more CpGs at a greater depth also increases confidence in the data and leads to more accurately defining methylation within a region of interest. This in turn aids in detecting methylation changes in diseased states such as cancer. In addition, increased CpG coverage has an economic impact – with more CpGs detected using the same number of reads compared to WGBS, EM-seq represents significant cost-savings.
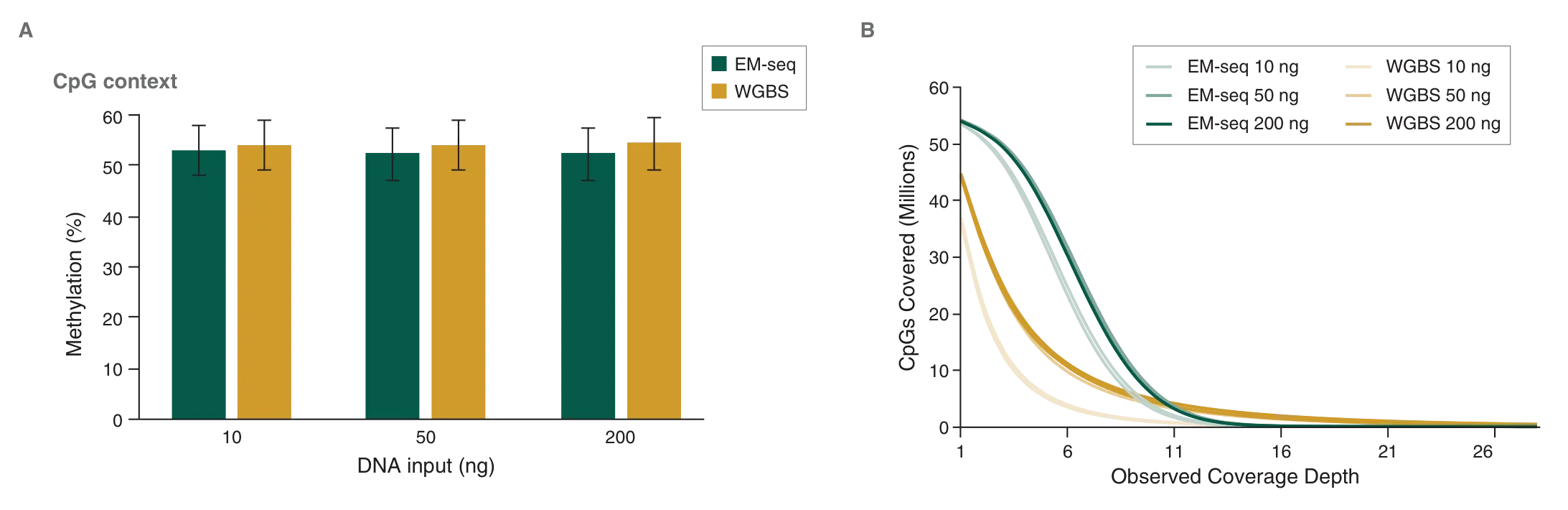
A: Methyl Dackel was used to determine methylation levels, which were found to be similar between EM-seq and WGBS.
B: Coverage of CpGs with EM-seq and WGBS libraries was analyzed, and each top and bottom strand CpGs were counted independently, yielding a maximum of 56 million possible CpG sites. EM-seq identifies more CpGs at lower depth of sequencing.
Potential Applications
In addition to making Illumina libraries, there are other potential applications for the EM-seq technology. Many of these applications already exist, but can now be improved upon because of the intact nature of enzymatically-converted DNA and the accuracy of CpG detection. Lower input DNA is also a driving factor for some of these applications. Converted DNA can be detected on arrays, and can be used for target enrichment, reduced representation-type libraries or amplicon detection. Different types of DNA inputs, such as low input cell free DNA (cfDNA) or damaged FFPE DNA, can also be used.
Conclusion
Bisulfite sequencing, while commonly used, is sub-optimal in detecting 5mC and 5hmC – large amounts of DNA are needed, DNA can be damaged, and sequences are biased towards AT-rich regions. Other methods that couple chemical or enzymatic treatment with bisulfite sequencing also share similar limitations. EM-seq provides the first commercially-available, non-bisulfite method that comprehensively addresses the limitations of bisulfite sequencing and represents a new opportunity for more complete methylome analysis. EM-seq libraries are not damaged and have longer inserts, higher PCR yields with fewer PCR cycles, and lack biases associated with GC content. More CpGs are identified with greater coverage depth using EM-seq, as compared to WGBS. These advantages all contribute to EM-seq having more usable sequencing data when comparing the same number of reads for EM-seq and WGBS, which ultimately reduces sequencing costs. EM-seq is the only commercially-available alternative to bisulfite sequencing that provides an effective method for accurate and comprehensive detection of 5mC and 5hmC across the genome, and offers a new, more accurate alternative for studying disease states.
References
- Harris R.A., et al. (2010) Nat Biotechnol. 28, 1097–1105.
- Olova, N., et al. (2018) Genome Biology, 19: 33.
- Yu, M., et al. (2012) Cell, 149, 1368–1380.
- Booth, M.J., et al. (2012) Science 336, 934–937.
- Schutsky, E.K., et al. (2018) Nature Biotechnology, 36, 1083–1090.
- Zhang, L., et al. (2013) Nat. Commun. 4: 1517.
- Carpenter, M.A., et al. (2012) J. Biol.Chem. 287, 34801–34808.
- Wijesinghe, P. and Bhagwat, A.S. (2012) Nucl. Acids Res. 40, 9206–9217.
- Josse, J and Kornberg, A. (1962) J. Biol. Chem. 237, 1968–1976.
- Tomaschewski, J., et al. (1985) Nucleic Acids Res.13 (21): 7551–7568.
- Schutsky, E.K., et al. (2017) Nucleic Acids Res. 45, 7655–7665.
