Breeding a better tomato with the NEBNext Direct® Genotyping Solution
By Andrew Barry, MS, New England Biolabs
The global population is anticipated to eclipse 8 billion by 2025, and approach 10 billion people by 2050 (1). Along with this rate of growth comes global challenges for feeding this population, pressing the need for more efficient farming practices. This efficiency is perturbed by reduced land availability for farming, emergence of novel pathogens, diminishing table water, and observed changes in climate, generating the need for sustainable crops with improved resilience to these stresses.
Traditional crop breeding approaches rely on interbreeding, or “crossing” of plant varieties for allelic transfer to generate genetic diversity within new crop varieties. This is followed by phenotypic assessment, and subsequent backcrossing with the parental lines for selection of desired traits. These traits can be quantitative and practical, such as pathogen resistance, drought tolerance and improvements in crop yield, or they can be more subjective and aesthetic traits including flavor and color. While these methods are effective, they rely on plant growth, exposure to stress, and observation of the desired phenotype in order to assess the presence of the desired trait; therefore, the breeding process is greatly lengthened.
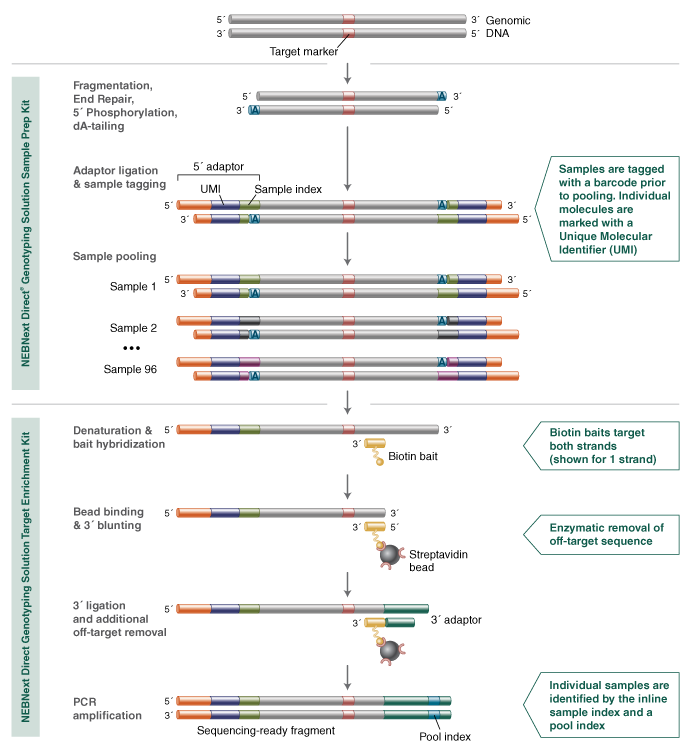
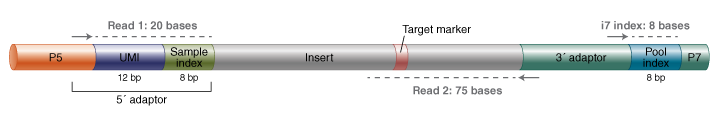
Figure 1B: Final library and sequencing details
Quantitative trait locus, or QTL mapping, generates linkage information between a desired phenotype and the associated genotypic information. There are several approaches available to perform QTL mapping, but the goal is to identify a set of genetic markers that can be used in place of phenotypic information to assess whether plants are harboring the specific markers that are positive indicators for the presence of traits being selected for. The development of crop-specific databases to guide breeding programs has created a need for novel methods for genotyping plants that result from genetic crossing or backcrossing in order to guide future breeding efforts. To generate necessary genetic diversity, thousands to tens of thousands of plants are used for breeding efforts, so ideally these approaches are fast and scalable to address the throughput demand.
Traditional genotyping approaches include endpoint PCR assays, whereby only a limited number of markers for any given plant can be assayed, and input samples must be split across multiple PCR reactions, requiring high amounts of PCR consumables and specialized equipment for high-throughput sample processing. Another common option for genotyping is the use of SNP-based microarrays, which can assay hundreds of thousands of markers in parallel, yet the challenge lies in scaling the experiment for high sample numbers, because a single DNA sample per chip is required. In parallel, the creation of full genome reference sequences for many crops has increased our knowledge, and therefore, additional marker types including genomic insertions and deletions, and combinations of markers, or haplotypes, are increasingly used as phenotypic indicators. Microarrays and endpoint PCR assays are limited in their ability to solely provide information on the presence or absence of a known marker and cannot be used for discovery of novel genomic information.
The advent of next-generation sequencing has provided scientists in many research areas with a tool to understand genomic information in a cost-effective manner. The continual decreases in the cost of sequencing have made this an attractive readout that provides not only genotype information, but contextual information of the areas surrounding target genomic loci, that can increase the types of available genomic markers, and also lead to the discovery of new markers. The efficiency of next-generation sequencing has shifted the throughput challenge further upstream, necessitating improved methods for preparing samples for sequencing analysis, in the most efficient way possible.
The NEBNext Direct Genotyping Solution was developed to address the specific needs of high-throughput, targeted genotyping required for breeding applications. Developed in collaboration with industrial crop breeders, the method employs high levels of sample multiplexing along with highly efficient, capture-based enrichment of hundreds to thousands of genomic loci, to enable next-generation sequencing to be fully leveraged as a means for high-throughput, cost-effective genotyping.
In order to demonstrate the efficacy of the approach, a panel of 2309 genomic markers was developed to target SNPs from the Solanaceae Coordinated Agricultural Project (SolCAP) database (2). This panel was assayed against extracted DNA from 96 samples of the tomato crop, Solanum lycopersicum, and processed using the NEBNext Genotyping Solution before Illumina® sequencing.
Key features aiding the efficiency of the NEBNext Direct Genotyping Solution include the consolidation of DNA fragmentation with end repair, 5´ phosphorylation, and dA-tailing into a single enzymatic step. This is immediately followed by ligation of the Sample Index, which contains a 5´ adaptor to barcode the samples. These two steps represent the only workflow steps where samples are processed individually, as sample pooling immediately follows. By pooling samples (up to 96) prior to capture-based enrichment, the processing steps are significantly reduced, and there is a vast reduction in laboratory consumables required. The 5´ adaptor also contains a 12 base pair, random sequence known as a Unique Molecular Identifier, or UMI. The UMI is used to individually index each library molecule and is used in data analysis to identify duplicate molecules that are generated during the downstream amplification processes, as well as aiding more accurate genotype calls.
The bait hybridization step hybridizes both DNA strands using synthetic, biotinylated oligonucleotides designed against all 2,309 genomic regions harboring the loci of interest for all 96 samples, capturing a total of over 220,000 data points in a single enrichment reaction. These captured molecules are subsequently fully converted into a next-generation sequencing library, during which specificity enhancing enzymatic treatments are performed, and a second, pool-specific barcode is added to the 3´ end of molecules. This dual-indexing strategy enables further pooling prior to sequencing, maximizing the output of Illumina® sequencing.
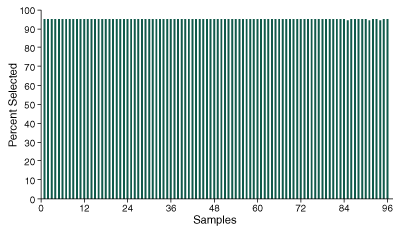
In order to assess performance across 96 samples, sequencing data was processed and aligned to the reference genome sequence, and key metrics were obtained. Analysis of the percent selected across each of the 96 samples demonstrates the method’s ability to efficiently enrich molecules containing the target SNP markers, with consistent values across samples showing > 95% of sequencing data mapping to the defined targets (Figure 2).
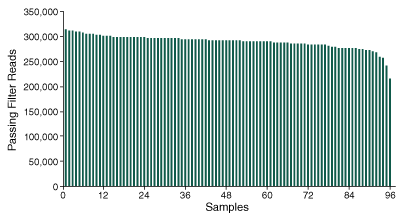
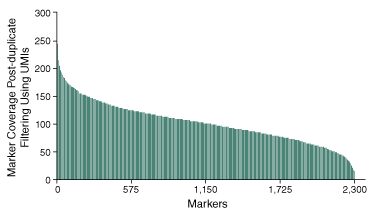
Further analysis into the ability to confidently determine the genotype of the 2,309 SNPs, demonstrates the ability of the method to produce consistent sequencing coverage at depths sufficient to assess the presence or absence of SNP markers across the 96 pooled samples (Figure 3).
A closer examination of the specific performance across targets included in the panel within a single sample shows coverage across the highest and lowest performing targeted regions within.
These data suggest that by using the NEBNext Direct Genotyping Solution, high-throughput, massively parallel enrichment of genomic loci can be achieved in an efficient manner upfront of next generation sequencing. The combined efficiency of a novel sample-preparation strategy and continued advances in next generation sequencing present a tractable solution for genotyping hundreds to thousands of genomic loci that can be employed to accelerate plant breeding programs aimed at production of new crop variants that can overcome the challenges our global population growth will continue to present.
View a PDF of this Feature Article
References:
- United Nations, Department of Economic and Social Affairs, Population Division (2019). World Population 2019: Wall Chart (ST/ESA/ SER.A/434).
- Solanaceae Coordinated Agricultural Project (SolCAP). 2011. Tomato intervarietal TA496 vs. Heinz1706 SNPs. Michigan State University, Dept. of Plant and Soil Science, East Lansing, MI. https://solcap.msu.edu.
- https://broadinstitute.github.io/picard
- https://solgenomics.net/help/index.pl
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN]
- Fulcrum Genomics, https://github.com/fulcrumgenomics/fgbio
