Protocol for use with NEBNext® UltraTM II RNA Library Prep Kit for Illumina® E7770 and rRNA Depletion Kit (Human/Mouse/Rat) (NEB #E6310)
 |
This is a point where you can safely stop the protocol and store the samples prior to proceeding to the next step in the protocol. |
 |
This caution sign signifies a step in the protocol that has two paths leading to the same end point but is dependent on a user variable, like the type of RNA input. |
 |
Colored bullets indicate the cap color of the reagent to be added |
This protocol has been optimized using high quality Universal Human Reference Total RNA.
RNA Sample Requirements
RNA Integrity:
Assess the quality of the input RNA by running the RNA sample on an Agilent Bioanalyzer RNA 6000 Nano/Pico Chip to determine the RNA Integrity Number (RIN). RNA with different RIN values require different fragmentation times or no fragmentation at all.
For intact (RIN > 7) or partially degraded RNA samples (RIN = 2 to 7) follow the library preparation protocol in Section 2 (current Section). See Table 2.5.1. for the recommended fragmentation times, based on RIN.
For highly degraded samples (RIN = 1 to 2) (e.g. FFPE), which do not require fragmentation, follow the library preparation protocol in Section 3.
RNA Sample Requirements:
The RNA sample should be free of salts (e.g., Mg2+, or guanidinium salts) or organics (e.g., phenol and ethanol). RNA must be free of DNA. gDNA is a common contaminant from RNA preps. It may be carried over from the interphase of organic extractions or when the silica matrix of solid phase RNA purification methods is overloaded. If the total RNA sample may contain gDNA contamination, treat the sample with DNase I to remove all traces of DNA (not provided in this kit). After treatment with DNase I the enzyme should be removed from the sample. Any residual activity of DNase I will degrade the single stranded DNA probes necessary for the ribosomal depletion. DNase I can be removed from the extraction using phenol/ chloroform extraction and ethanol precipitation.
Input Amount Requirement
5 ng–1 μg total RNA (DNA free) in a 12 μl total volume, quantified by Qubit Fluorometer and quality checked by Bioanalyzer. The protocol is optimized for approximately 200 nt RNA inserts. To generate libraries with longer RNA insert sizes, refer to Appendix A (Section 6) for recommended fragmentation times and size selection conditions.
Keep all of the buffers on ice, unless otherwise indicated.
2.1. Probe Hybridization to RNA
2.1.1. Dilute the total RNA with Nuclease-free Water to a final volume of 12 μl in a PCR tube. Keep the RNA on ice.
2.1.2. Prepare a RNA/Probe master mix as follows:
| RNA/PROBE MASTER MIX | VOLUME |
|---|---|
| NEBNext rRNA Depletion Solution | 1 µl |
| Probe Hybridization Buffer |
2 µl |
| Total Volume |
3 µl |
2.1.3. Add 3 µl of the above mix to 12 µl total RNA (from Step 2.1.1), resulting in a total volume of 15 µl.
2.1.4. Mix thoroughly by pipetting up and down at least 10 times.
2.1.5. Briefly spin down the sample in a microcentrifuge.
2.1.6. Place samples in a thermocycler, and run the following program with the heated lid set at 105°C. This will take approximately 15-20 minutes to complete.
| TEMPERATURE | TIME |
|---|---|
| 95°C | 2 minutes |
| Ramp down to 22°C |
0.1°C/sec |
| 22°C |
5 minutes |
2.1.7. Briefly spin down the sample in a microcentrifuge, and place on ice. Proceed immediately to RNase H Digestion Step.
2.2. RNase H Digestion
2.2.1. Assemble the RNase H master mix on ice as follows.
| RNASE H MASTER MIX | VOLUME |
|---|---|
| NEBNext RNase H | 2 µl |
| NEBNext RNase H Reaction Buffer |
2 µl |
| Nuclease-free Water |
1 µl |
| Total Volume |
5 µl |
2.2.2. Mix thoroughly by pipetting up and down at least 10 times.
2.2.3. Briefly spin down the samples in a microcentrifuge.
2.2.4. Add 5 µl of the RNase H master mix to the RNA sample from Step 2.1.7, resulting in a total volume of 20 µl.
2.2.5. Mix thoroughly by pipetting up and down at least 10 times.
2.2.6. Incubate the sample in a thermocycler for 30 minutes at 37°C with the lid set to 40°C (or on).
2.2.7. Briefly spin down the samples in a microcentrifuge, and place on ice. Proceed immediately to DNase I Digestion to prevent non-specific degradation of RNA.
2.3. DNase I Digestion
2.3.1. Assemble the DNase I master mix on ice in a nuclease-free tube.
| DNASE I MASTER MIX | VOLUME |
|---|---|
| DNase I Reaction Buffer | 5 µl |
| DNase I (RNase-free) |
2.5 µl |
| Nuclease-free Water |
22.5 µl |
| Total Volume |
30 µl |
2.3.2. Mix thoroughly by pipetting up and down at least 10 times.
2.3.3. Briefly spin down the sample in a microcentrifuge.
2.3.4. Add 30 μl of DNase I master mix to 20 μl RNA sample from Step 2.2.7, resulting in a total volume of 50 μl.
2.3.5. Mix thoroughly by pipetting up and down ten times.
2.3.6. Incubate the sample in a thermocycler for 30 minutes at 37°C with the heated lid set to 40°C (or on).
2.3.7. Briefly spin down the sample in a microcentrifuge, and place on ice. Proceed immediately to RNA Purification.
2.4 RNA Purification Using Agencourt RNAClean® XP Beads or NEBNext RNA Sample Purification Beads
2.4.1. Vortex the RNAClean XP or RNA Sample Purification Beads to resuspend.
2.4.2. Add 110 µl (2.2X) beads to the RNA sample from Step 2.3.7 and mix thoroughly by pipetting up and down at least 10 times.
2.4.3. Incubate the sample for 15 minutes on ice to bind RNA to the beads.
2.4.4. Place the tube on a magnetic rack to separate beads from the supernatant. After the solution is clear, carefully remove and discard the supernatant. Be careful not to disturb the beads, which contain RNA.
2.4.5. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the RNA.
2.4.6. Repeat Step 2.4.5 once for a total of 2 washing steps.
2.4.7. Completely remove residual ethanol, and air dry the beads for up to 5 minutes while the tube is on the magnetic rack with the lid open.
Caution: Do not over-dry the beads. This may result in lower recovery of RNA target. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack they are too dry.
2.4.8. Remove the tube from the magnet. Elute the RNA from the beads by adding 7 µl Nuclease-free Water. Mix well by pipetting up and down at least 10 times and briefly spin the tube.
2.4.9. Incubate for 2 minutes at room temperature. Place the tube in the magnet until the solution is clear (~2 minutes).
2.4.10. Remove 5 µl of the supernatant containing RNA and transfer to a nuclease-free tube.
2.4.11. Place the sample on ice and proceed to RNA Fragmentation and Priming.
2.5. RNA Fragmentation and Priming
RNA fragmentation is only required for intact or partially degraded RNA. Recommended fragmentation times can be found in Table 2.5.1. 2.5.1. Assemble the following fragmentation and priming reaction on ice:
| FRAGMENTATION AND PRIMING REACTION |
VOLUME |
|---|---|
| Ribosomal RNA Depleted Sample (Step 2.4.11) | 5 µl |
 (lilac) NEBNext First Strand Synthesis Reaction Buffer (lilac) NEBNext First Strand Synthesis Reaction Buffer |
4 µl |
| (lilac) Random Primers |
1 µl |
| Total Volume |
10 µl |
2.5.2. Mix thoroughly by pipetting up and down ten times.
2.5.3. Place the sample on a thermocycler and incubate the sample at 94°C following the recommendations in Table 2.5.1 below for libraries with inserts sizes ~200 nt.
Table 2.5.1. Suggested fragmentation times based on RIN value of RNA input.
| RNA TYPE | RIN | FRAG. TIME |
|---|---|---|
| Intact RNA | > 7 | 15 min. at 94°C |
| Partially Degraded RNA | 2–6 | 7–8 min. at 94°C |
Note: Refer to Appendix A (Section 6) for fragmentation conditions if you are preparing libraries with large inserts (> 200 bp). Conditions in Appendix A only apply for intact RNA.
2.5.4. Immediately transfer the tube to ice and proceed to First Strand cDNA Synthesis.
2.6. First Strand cDNA Synthesis
2.6.1. Assemble the first strand synthesis reaction on ice by adding the following components to the fragmented and primed RNA from Step 2.5.4:
| FIRST STRAND SYNTHESIS REACTION | VOLUME |
|---|---|
| Fragmented and Primed RNA (Step 2.5.4) | 10 µl |
| Nuclease-free Water |
8 µl |
| (lilac) NEBNext First Strand Synthesis Enzyme Mix |
2 µl |
| Total Volume |
20 µl |
2.6.2. Mix thoroughly by pipetting up and down ten times.
2.6.3.
Incubate the sample in a preheated thermocycler with the heated lid set at ≥ 80°C as follows:Note: If you are following recommendations in Appendix A (Section 6), for libraries with longer inserts (> 200 bases), increase the incubation at 42°C from 15 minutes to 50 minutes at Step 2 below.
Step 1: 10 minutes at 25°C
Step 2: 15 minutes at 42°C
Step 3: 15 minutes at 70°C
Step 4: Hold at 4°C
2.7. Second Strand cDNA Synthesis
2.7.1. Assemble the second strand cDNA synthesis reaction on ice by adding the following components into the first strand synthesis product from Step 2.6.4.
| SECOND STRAND SYNTHESIS REACTION | VOLUME |
|---|---|
| First Strand Synthesis Product (Step 2.6.4) | 20 µl |
 (orange) NEBNext Second Strand Synthesis Reaction Buffer (10X) (orange) NEBNext Second Strand Synthesis Reaction Buffer (10X) |
8 µl |
| (orange) NEBNext Second Strand Synthesis Enzyme Mix |
4 µl |
| Nuclease-free Water |
48 µl |
| Total Volume |
80 µl |
2.7.2. Keeping the tube on ice, mix thoroughly by pipetting up and down at least 10 times.
2.7.3. Incubate in a thermocycler for 1 hour at 16°C with the heated lid set at ≤ 40°C (of off).
2.8. Purification of Double-stranded cDNA Using SPRIselect Beads or NEBNext Sample Purification Beads
2.8.1. Vortex SPRIselect beads or NEBNext Sample Purification Beads to resuspend.
2.8.2. Add 144 μl (1.8X) of resuspended beads to the second strand synthesis reaction (~80 μl). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
2.8.3. Incubate for 5 minutes at room temperature.
2.8.4.Briefly spin the tube in a microcentrifuge to collect any sample on the sides of the tube. Place the tube on a magnet to separate beads from the supernatant. After the solution is clear, carefully remove and discard the supernatant. Be careful not to disturb the beads, which contain DNA. (Caution: do not discard beads).
2.8.5. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
2.8.6. Repeat Step 2.8.5 once for a total of 2 washing steps.
2.8.7. Air dry the beads for up to 5 minutes while the tube is on the magnetic rack with lid open.
Caution: Do not over-dry the beads. This may result in lower recovery of DNA target. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack they are too dry.
2.8.8. Remove the tube from the magnetic rack. Elute the DNA from the beads by adding 53 μl 0.1X TE Buffer (provided) to the beads. Mix well on a vortex mixer or by pipetting up and down at least 10 times. Quickly spin the tube and incubate for 2 minutes at room temperature. Place the tube on the magnetic rack until the solution is clear.
2.8.9. Remove 50 µl of the supernatant and transfer to a clean nuclease-free PCR tube.
| If you need to stop at this point in the protocol, samples can be stored at –20°C. |
2.9. End Prep of cDNA Library
2.9.1. Assemble the end prep reaction on ice by adding the following components to the second strand synthesis product from Step 2.8.9.
| END PREP REACTION | VOLUME |
|---|---|
| Second Strand Synthesis Product (Step 2.8.9) | 50 µl |
| (green) NEBNext Ultra II End Prep Reaction Buffer |
7 µl |
| (green) NEBNext Ultra lI End Prep Enzyme Mix |
3 µl |
| Total Volume |
60 µl |
2.9.2. Set a 100 μl or 200 μl pipette to 50 μl and then pipette the entire volume up and down at least 10 times to mix thoroughly. Perform a quick spin to collect all liquid from the sides of the tube.
Note: It is important to mix well. The presence of a small amount of bubbles will not interfere with performance.
2.9.3. Incubate the sample in a thermal cycler with the heated lid set at ≥ 75°C as follows.
30 minutes at 20°C
30 minutes at 65°C
Hold at 4°C.
2.9.4. Proceed immediately to Adaptor Ligation.
2.10. Adaptor Ligation
2.10.1. Dilute the  (red) NEBNext Adaptor* prior to setting up the ligation reaction in ice-cold Adaptor Dilution Buffer and keep the adaptor on ice.
(red) NEBNext Adaptor* prior to setting up the ligation reaction in ice-cold Adaptor Dilution Buffer and keep the adaptor on ice.
| TOTAL RNA INPUT | DILUTION REQUIRED |
|---|---|
| 1,000 ng–101 ng | 5-fold dilution in Adaptor Dilution Buffer |
| 100 ng–10 ng |
25-fold dilution in Adaptor Dilution Buffer |
| 5 ng |
200-fold dilution in Adaptor Dilution Buffer |
*The NEBNext adaptor is provided in NEBNext oligos kit. NEB has several oligo kit options, which are supplied separately from the library prep kit.
2.10.2. Assemble the ligation reaction on ice by adding the following components, in the order given, to the end prep reaction product from Step 2.9.4.
| LIGATION REACTION | VOLUME |
|---|---|
| End Prepped DNA (Step 2.9.4) | 60 µl |
| Diluted Adaptor (Step 2.10.1) |
2.5 µl |
 (red) NEBNext Ligation Enhancer (red) NEBNext Ligation Enhancer |
1 µl |
| (red) NEBNext Ultra II Ligation Master Mix |
30 µl |
| Total Volume |
93.5 µl |
Note: The Ligation Master Mix and Ligation Enhancer can be mixed ahead of time and is stable for at least 8 hours @ 4°C. We do not recommend premixing the Ligation Master Mix, Ligation Enhancer and adaptor prior to use in the Adaptor Ligation Step.
2.10.3. Set a 100 μl or 200 μl pipette to 80 μl and then pipette the entire volume up and down at least 10 times to mix thoroughly. Perform a quick spin to collect all liquid from the sides of the tube.
| Caution: The NEBNext Ultra II Ligation Master Mix is very viscous. Care should be taken to ensure adequate mixing of the ligation reaction, as incomplete mixing will result in reduced ligation efficiency. The presence of a small amount of bubbles will not interfere with performance. |
2.10.4. Incubate 15 minutes at 20°C in a thermocycler.
2.10.5. Add 3 μl
 (red) USER Enzyme to the ligation mixture from Step 2.10.4, resulting in total volume of 96.5 μl.
(red) USER Enzyme to the ligation mixture from Step 2.10.4, resulting in total volume of 96.5 μl. 2.10.6. Mix well and incubate at 37°C for 15 minutes with the heated lid set to ≥ 45°C.
2.10.7. Proceed immediately to Purification of the Ligation Reaction.
2.11. Purification of the Ligation Reaction Using SPRIselect Beads or NEBNext Sample Purification Beads
| If you are selecting for libraries with larger insert size (> 200 nt) follow the size selection recommendations in Appendix A, Section 6. |
2.11.1. Add 87 μl (0.9X) resuspended SPRIselect Beads or NEBNext Sample Purification Beads and mix well on a vortex mixer or by pipetting up and down at least 10 times.
2.11.2. Incubate for 5 minutes at room temperature.
2.11.3. Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from the supernatant. After the solution is clear (~ 5 minutes), discard the supernatant that contains unwanted fragments. Caution: do not discard the beads.
2.11.4. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
2.11.5. Repeat Step 2.11.4 once for a total of 2 washing steps.
2.11.6. Briefly spin the tube, and put the tube back in the magnetic rack.
2.11.7. Completely remove the residual ethanol, and air dry beads until the beads are dry for up to 5 minutes while the tube is on the magnetic rack with the lid open.
Caution: Do not over-dry the beads. This may result in lower recovery of DNA target. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack they are too dry.
2.11.8. Remove the tube from the magnetic rack. Elute DNA target from the beads by adding 17 μl 0.1X TE (provided) to the beads. Mix well on a vortex mixer or by pipetting up and down. Quickly spin the tube and incubate for 2 minutes at room temperature. Put the tube in the magnet until the solution is clear.
2.11.9. Without disturbing the bead pellet, transfer 15 μl of the supernatant to a clean PCR tube and proceed to PCR enrichment.
| If you need to stop at this point in the protocol, samples can be stored at –20°C. |
2.12. PCR Enrichment of Adaptor Ligated DNA
Check and verify that the concentration of your oligos is 10 μM on the label.
Use Option A for any NEBNext oligos kit where index primers are supplied in tubes. These kits have the forward and reverse primers supplied in separate tubes.
Use Option B for any NEBNext oligos kit where index primers are supplied in a 96-well plate format. These kits have the forward and reverse (i7 and i5) primers combined.
2.12.1. Set up the PCR reaction as described below based on the type of oligos (PCR primers) used.
2.12.1A Forward and Reverse Primers Separate
| COMPONENT | VOLUME PER ONE LIBRARY |
|---|---|
| Adaptor Ligated DNA (Step 2.11.9) | 15 µl |
 (blue) NEBNext Ultra II Q5 Master Mix (blue) NEBNext Ultra II Q5 Master Mix |
25 µl |
| Index (X) Primer /i7 Primer*,** |
5 µl |
| Universal PCR Primer/i5 Primer*, ** |
5 µl |
| Total Volume |
50 µl |
* NEBNext Oligos must be purchased separately from the library prep kit. Refer to the corresponding NEBNext Oligo kit manual for determining valid barcode combinations.
** Use only one i7 primer/ index primer per sample. Use only one i5 primer (or the universal primer for single index kits) per sample
2.12.1B Forward and Reverse Primers Combined
| COMPONENT | VOLUME PER ONE LIBRARY |
|---|---|
| Adaptor Ligated DNA (Step 2.11.9) | 15 µl |
| (blue) NEBNext Ultra II Q5 Master Mix |
25 µl |
| Index Primer Mix* |
10 µl |
| Total Volume |
50 µl |
* NEBNext Oligos must be purchased separately from the library prep kit. Refer to the corresponding NEBNext Oligo kit manual for determining valid barcode combinations.
2.12.2. Mix well by gently pipetting up and down 10 times. Quickly spin the tube in a microcentrifuge.
2.12.3. Place the tube on a thermocycler with the heated lid set to 105°C and perform PCR amplification using the following PCR cycling conditions (refer to Table 2.12.3A and Table 2.12.3B):
Table 2.12.3A:
| CYCLE STEP |
TEMP | TIME |
CYCLES |
|---|---|---|---|
| Initial Denaturation | 98°C |
30 seconds | 1 |
| Denaturation Annealing/Extension |
98°C 65°C |
10 seconds 75 Seconds |
6–15*, ** |
| Final Extension | 65°C |
5 minutes | 1 |
| Hold | 4°C |
∞ |
*The number of PCR cycles should be adjusted based on RNA input.
**It is important to limit the number of PCR cycles to avoid overamplification.
If overamplification occurs, a second peak ~ 1,000 bp will appear on the Bioanalyzer trace (See Figure 7.2 Appendix A).
Table 2.12.3B: Recommended PCR cycles based on total RNA input amount:
| TOTAL RNA INPUT | RECOMMENDED PCR CYCLES |
|---|---|
| 1,000 ng | 6–7 |
| 100 ng |
10–11 |
| 10 ng | 13–14 |
| 5 ng |
14–15 |
Note: PCR cycles are recommended based on high quality Universal Human Reference Total RNA. It may require optimization based on the sample quality to prevent PCR over-amplification.
2.13. Purification of the PCR Reaction using SPRIselect Beads or NEBNext Sample Purification Beads
2.13.1. Vortex SPRIselect Beads or NEBNext Sample Purification Beads to resuspend.
2.13.2. Add 45 μl (0.9X) of resuspended beads to the PCR reaction (~ 50 μl). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
2.13.3. Incubate for 5 minutes at room temperature.
2.13.4. Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from the supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets. (Caution: do not discard beads).
2.13.5. Add 200 μl of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
2.13.6. Repeat Step 2.13.5 once for a total of 2 washing steps.
2.13.7. Air dry the beads for up to 5 minutes while the tube is on the magnetic rack with the lid open.
Caution: Do not over-dry the beads. This may result in lower recovery of DNA target. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack they are too dry.
2.13.8. Remove the tube from the magnetic rack. Elute the DNA target from the beads by adding 23 μl 0.1X TE (provided) to the beads. Mix well on a vortex mixer or by pipetting up and down ten times. Quickly spin the tube in a microcentrifuge and incubate for 2 minutes at room temperature. Place the tube in the magnetic rack until the solution is clear.
2.13.9. Transfer 20 μl of the supernatant to a clean PCR tube, and store at –20°C.
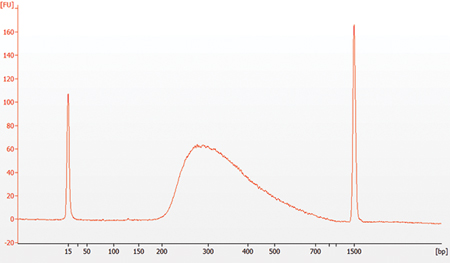
2.14. Assess Library Quality on an Agilent Bioanalyzer DNA Chip
2.14.1. Run 1 μl library on a DNA 1000 chip. If the library yield is too low to quantify on this chip, please run the samples on a DNA High Sensitivity chip. A dilution may be necessary for running on a Bioanalyzer High Sensitivity DNA Chip.
2.14.2. Check that the electropherogram shows a narrow distribution with a peak size approximately 300 bp.
Note: If a peak at ~ 80 bp (primers) or 128 bp (adaptor-dimer) is visible in the bioanalyzer traces, bring up the sample volume (from Step 2.13.9) to 50 μl with 0.1X TE buffer and repeat the SPRIselect Bead Cleanup Step (Section 2.13).