NEB® TV Ep. 21 - RNA modifications
Script
NEB TV, what's trending in science.
Deana Martin:
Welcome to NEB TV. Today I am joined by Joe Whipple, who is one of our application and development scientists, hey Joe!
Joe Whipple
Hi Deana.
Deana Martin:
And today we're talking about RNA modifications.
In our science in 60, Joe will be providing an overview of the different types of RNA modifications that exist. Next, we will hear from NEB scientist Laurence Etwiller, and she will be talking about cappable-seq. Then we will hear from Thomas Begley, who is the Interim Director of the RNA institute at The University of Albany and he will be providing an overview of the RNA Database. Lastly, we will hear from Ivan Correa, an NEB scientist who will be talking about the Nucleoside Digestion Mix, which can be used for the detection and characterization of DNA or RNA modifications.
Are you ready?
Joe Whipple:
I am.
Deana Martin:
Alright, let's get started.
Science in 60
Joe Whipple:
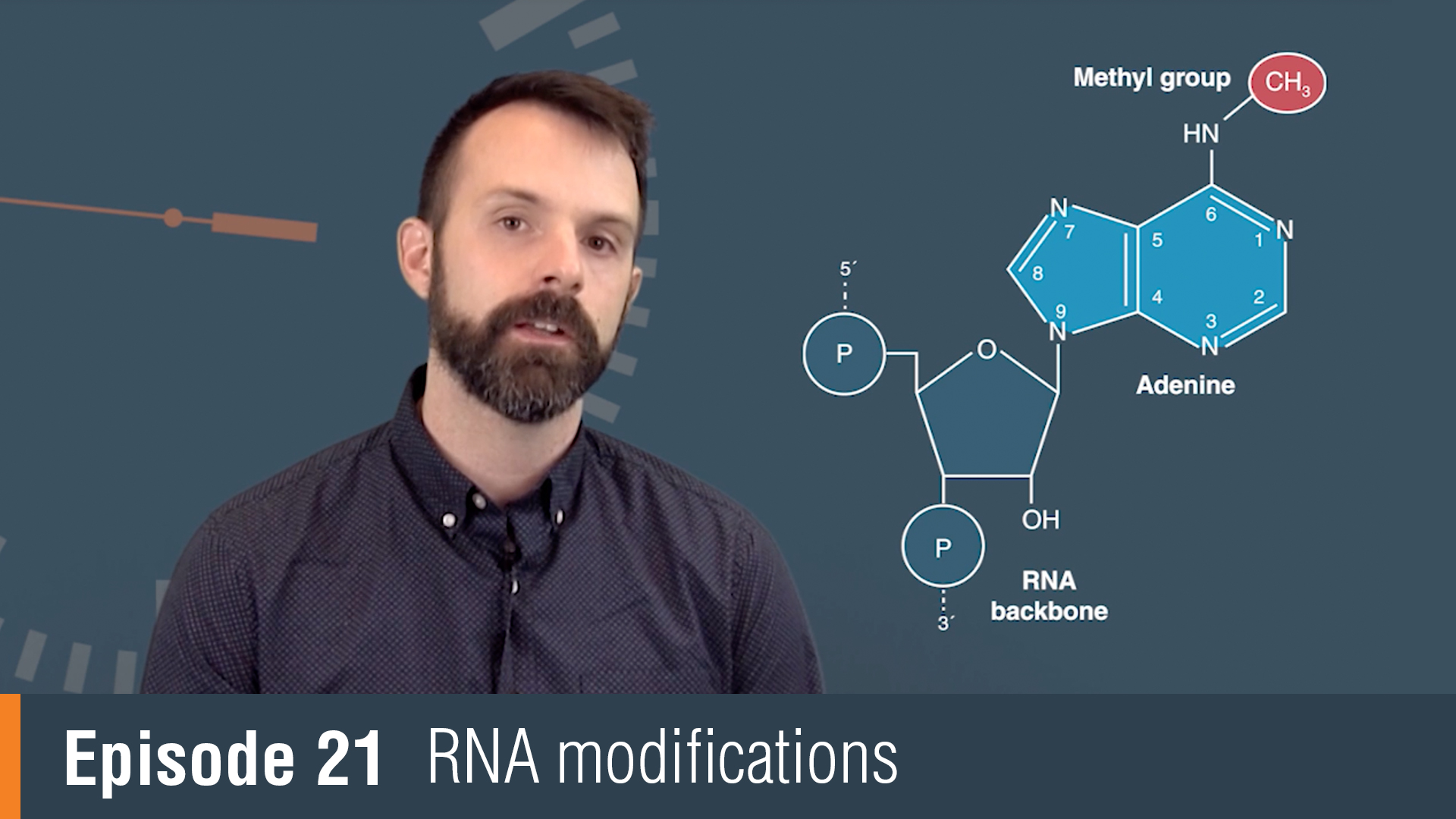
RNA modifications or collectively, the epitranscriptome encompass all chemical changes made to ribonucleic acids during and after synthesis and their influence extends from the bench to the clinic. RNA modifications were originally discovered in the most abundant cellular transcripts. tRNAs in particular had been a rich source of modifications with over 100 modifications discovered across all studied organisms to date. Much of what we know about the function of RNA modifications actually comes from the study of tRNA.
For example, tRNA research has shown us how modifications influence base pairing, stability and function. With the advent of more sensitive detection techniques, RNA modifications have been found on nearly all cellular transcripts, including mRNA. N6-Methyladenosine, or M6A, has been shown to be a dynamic modification that's important for timing RNA translation. ADARs or adenosine deaminases, act exclusively on double-stranded RNA and play a critical function. Adenosine deamination in coding regions can change the coding potential of mRNA and expand the number of proteins that one gene can encode. RNA modifications can also influence how our bodies innate immune system responds to foreign RNA. 2'-O-methylation for example can mask foreign RNA from our body's immune system.
Cappable-seq
Laurence Etwiller:
Cappable-seq explains the fact that primary transcript prokaryotics have a unique triphosphate at the beginning of the RNA. Cappable-seq uses this feature to capture the 5´ end of this molecule, enabling determination of transcription start site at base resolution. In addition to the TSS determination, Cappable-seq depletes ribosomal RNA and reduces the complexity of the transcriptome to a single quantifiable tag per transcript, enabling digital profiling of gene expression in a microbiome.
Are there other methods to identify transcription start sites?
For prokaryotes, tRNA-seq has been used extensively to identify transcription start site. It uses the terminator exonuclease TEX treatment to vertically remove all non- triphosphate RNA. The limitation of this technology is that success seems to highly depend on the 5´ end structure of the RNA. Therefore, the removal of non-triphosphate RNA, so the non TSS, is incomplete and in some cases nonexistent, leading to false positive TSS. For example, tRNA and large ribosomal contamination.
The advantage of Cappable-seq over the TEX method is the direct capturing of the triphosphate as opposed to the depletion of the non-triphosphate, which leads to a higher specificity, removal of the ribosomal RNA, and improved tolerance for low quality RNA.
What is SMRT-Cappable-seq?
What are the key differences and what kinds of biological questions can be studied with this technique?
We recently reported* about SMRT-Cappable-Seq, a derivative of Cappable-Seq that sequence full length RNA using PacBio as opposed to Cappable-Seq that identifies transcription start site only.
We applied SMRT Cappable-Seq to the best annotative prokaryotic genome, E. coli, and demonstrated 34 percent of the operon in E. coli are longer than the known annotation by at least one additional gene.
Phasing operon also enabled to observe operon variants and the granularity in the operon structure of E. coli that is reminiscent of prokaryotic splicing. SMRT-Cappable-Seq therefore describes a complete genomewide landscape of bacteria isoform. Similar to Cappable-Seq, SMRT Cappable-Seq also significantly remove ribosomal RNA.
The RNA Database
Hi I'm Tom Begley, Interim Director of the RNA Institute at The University of Albany here to tell you about the RNA modification database, maintained and updated at the RNA Institute.
The RNA modification database was started by Jim McCloskey, Pam Crain, and Jeff Rozenski many decades ago and continued by Paul Agris when the RNA Institute opened in 2010.
Currently there are 112 chemically verified modified nucleosides in the database, some simple, some complex, but all chemically verified. 11-plus RNA sources are represented in the database and include mRNA, tRNA, rRNA, snRNA, and many others.
In addition, RNA from archaea, bacteria, and eukaryea are found in the database. The modification database provides information about the chemical structure and the verifiable chemistry that was conducted. Researchers can look up the various RNAs for modifications starting with well-known bases U, A, I, C, and G, or go to specific modifications like my favorites M6A and MCM5U.
Researchers also benefit from search options, like filtering modifications based on the parent nucleosides to find the structure and chemistry of modifications that they are looking for. When we come across a publication about a new modification, or are contacted by the office of a publication detailing the structure and chemistry of the modification along with its natural occurrence, we add the modification to the database.
To help us enter new modifications, researchers should go to the database website and follow the contact information. We would appreciate a detailed message.
Importantly, the RNA modification database is accompanied by UNAFold, the most used and trusted secondary restructure program for folding of RNAs. Modified nucleosides will enter UNAFold when we have the force field parameters so that their influence on RNA folding can be assessed. We look forward to having you use the RNA Modification Database.
Identification and Quantitation of RNA Modifications
Ivan Correa:
The Nucleoside Digestion Mix is an optimized mixture of enzymes for our convenient one-step method to generate single nucleosides for quantitative analysis of DNA or RNA modifications. It digests single, double stranded, or DNA and RNA hybrids and tolerates a wide range of base and ribose modifications.
In the first step, genomic or synthetic DNA or RNA is converted into individual nucleotides by the mixed nuclease activity. In the second step, the nucleotides are dephosphorylated by the action of an unspecific phosphatase.
Here, I'd like to highlight an example where the Nucleoside Digestion Mix was used to monitor the metabolic incorporation of zero modified nucleosides into cellular RNA. Total RNA from cells treated with six ethylazido, six propylazido, and 2 azido adenosine was digested to ribonucleosides using the Nucleoside Digestion Mix and analyzed by LC-MS. The digested RNA from wild-type cells is shown in red. The insert shows an expansion of the overlaid chromatograms demonstrating the specific detection of the chemically modified nucleosides.
To learn more, please visit the Nucleoside Digestion Mix product page on NEB.com and download our latest application note.
Deana Martin:
So Joe, thanks so much for joining us today.
Joe Whipple:
No problem Deana.
Deana Martin:
And thank you for watching, if you have any suggestions for future episodes please let us know.
(*Yan, B. et al. (2018) bioRxiv, doi.org/10.1101/262964 Now published in Nature Communications, doi: 10.1038/s41467-018-05997-6)
Related Videos
-

In Vitro Transcription and Capping of Gaussia Luciferase mRNA Followed by HeLa Cell Transfection -

What is Cappable-seq? -
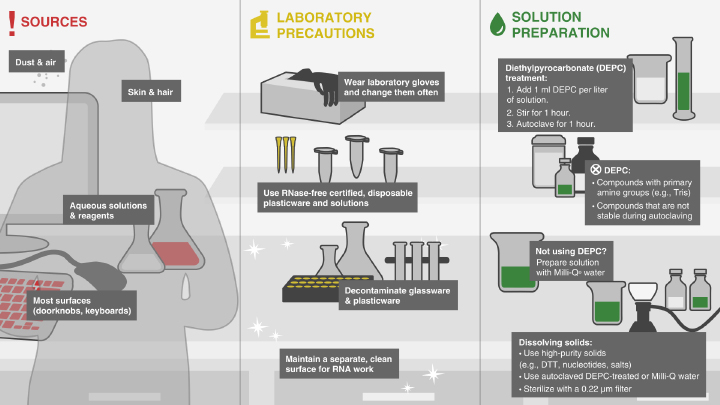
Avoiding RNase Contamination

