The Quantitation Question: How does accurate library quantitation influence sequencing?
The determination of the number of sequencing-ready molecules present after library preparation is an important step in the next generation sequencing (NGS) workflow and has a strong influence on the success of both a sequencing run and a sequencing-based experiment. Before selecting the quantitation method you’ll use, it can be helpful to understand what happens to a library during sequencing, and exactly what quantitation does and does not tell you. A typical NGS workflow starts with library preparation and ends with sequencing and data analysis; each of these steps is of critical importance to the quality and reproducibility of the sequencing data. However, between library preparation and sequencing is, perhaps, one of the less-discussed steps in the NGS workflow: library quantitation.
Why Do I Need to Quantitate My Library?
There are two primary reasons that libraries must be quantitated.
- The chemistries that underlie Illumina sequencing require an optimal amount of adaptor-ligated DNA fragments to be loaded into the cluster generation step, for example 6-10 pM for the MiSeq® instrument (v3 chemistry).
- If multiple libraries are sequenced in one run, it is desirable for the sequence coverage to be equal for each library, and therefore an equal amount of each library should be moved into the cluster generation step.
What happens to your library during sequencing?
To fully understand the importance of accurate library quantitation before sequencing, it is first necessary to understand sequencing chemistries and their interactions with the samples you’ll be sequencing.
For the purposes of this article, we’ll focus on the chemistries that underlie the popular (and market leading) Illumina sequencers, although library quantitation is an important step for sequencing on any platform.
Building bridges & counting clusters
Core components of Illumina’s sequencing tech- nology are its flow cells and their cluster-generating capabilities. Illumina’s sequencers are based on optical detection of DNA clusters that form on the glass flow cell, a phenomenon enabled by a dense lawn of primers pre-immobilized to the flow cell channel. As you add your library to the flow cell, the single-stranded, adaptor-ligated fragments hybridize to the immobilized primers studded across the flow cell. This step is where the accuracy of your library quantitation is put to the test.
Cluster generation then occurs: each hybridized molecule undergoes multiple rounds of amplification to produce up to 1,000 copies of the same molecule in the same location on the flow cell: a “cluster”, whose diameter is 1 micron or less. For more details on cluster generation, visit Illumina.com.
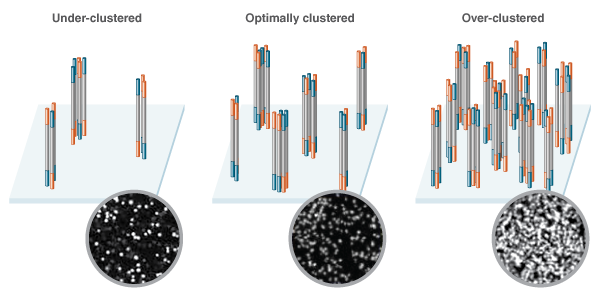
The amount of DNA initially loaded onto the flow cell directly influences the density of the clusters that form. Too little DNA and the clusters are likely to sparsely populate the flow cell. Too much DNA and the clusters will be too close together, making it difficult to interpret the sequencing data due to poor resolution, and resequencing of libraries will be required (Figure 1). Illumina’s recommended input ranges, which differ depending on the specific Illumina instrument, help to ensure that the clusters forming on the flow cell have sufficient resolution, without wasting valuable flow cell space.
The density of library clusters as they form on the flow cell prior to sequencing is a key factor in the success of a sequencing run. Low concen- tration libraries (Left) fail to make optimal use of the space, while high concentration libraries (Right) lead to densely packed clusters that are difficult to call. Optimal cluster density (Center) makes the best use of flow cell real estate, without over crowding. Representative optical data generated during sequencing depicts variation in cluster densities as shown in the insets.
A deeper dive into equivalent representation
When you pool libraries, you increase the value of each sequencing run by increasing the number of samples that can be sequenced in a single run. However, if libraries are combined in unequal concentrations, this leads to biased representation of certain libraries over oth- ers. In cases where libraries are significantly under-represented, these libraries will need to be resequenced, costing time and money. Over-rep- resentation of libraries can result in generation of more sequence data than required, and the subsequent discarding of sequence reads, wasting sequence capacity.
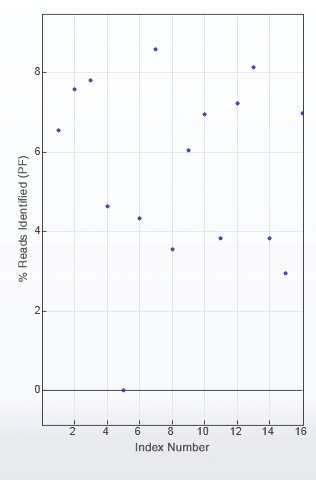
Figure 2 is an example of uneven library pooling resulting in uneven sequence coverage and the need to resequence. With 16 libraries in this pool, each library should theoretically have 6.25% of the sequence reads. However, this is not the case, and some of the libraries, such as libraries 5 and 15, would need to be resequenced.
Inadequate or uneven pooling of libraries can result in suboptimal data, and even lead to the need for library resequencing, as seen with library #5.
Why do my library fragments need to be adaptor-ligated?
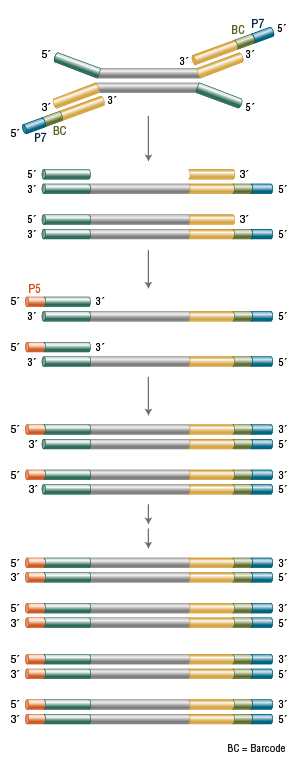
Optimal cluster density enables efficient & accurate quantitation The density of library clusters as they form on the flow cell prior to sequencing is a key factor in the success of a sequencing run. Low concen- tration libraries (Left) fail to make optimal use of the space, while high concentration libraries (Right) lead to densely packed clusters that are difficult to call. Optimal cluster density (Center) makes the best use of flow cell real estate, without over crowding. Representative optical data generated during sequencing depicts variation in cluster densities as shown in the insets. Sequences required downstream of library preparation, such as those for cluster generation and sequencing, must be added to the DNA fragments to be sequenced, and this is the primary goal of library preparation. In PCR-free library preparation workflows, all of the required sequences must be included in the adaptor sequence. In workflows including amplification, some of the sequences, including the sequences required for cluster generation (indicated by P5 and P7 in Figure 3,), can be added during PCR instead.
The stepwise addition of the sequences P5 and P7 and the barcode (BC) can be achieved during PCR amplification of the library.
Only fragments that have a P5 sequence at one end and a P7 sequence at the other are capable of participating successfully in cluster generation. Therefore, ideally, only fragments to which both of these sequences have been attached should be counted during a library quantitation step.
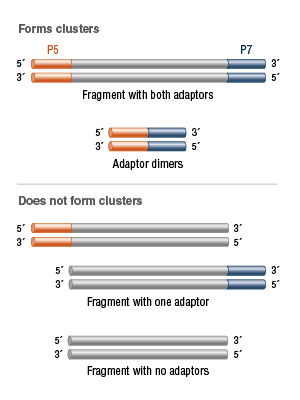
However, in addition to the desired fragments with an adaptor at both ends, libraries may also contain fragments that have no adaptors, one adaptor or adaptor-dimers. Fragments with no adaptors or one adaptor ligated will not form clusters. Adaptor-di- mers will efficiently cluster, but contain no DNA of interest (Figure 4).
Only library fragments containing both a P5 and a P7 adaptor will result in a flow-cell cluster. Other molecules are insufficient for cluster formation or contain no DNA of interest, so efforts should be made to exclude them from quantitation.
How Should I Quantitate My DNA Library?
Simply put, library quantitation refers to a variety of methods for determining the number of nucleic acid molecules present in a specific volume of your library. Unlike other molecular biology techniques, where the recommended input range is broad and forgiving, the basic chemistry of NGS requires that a narrow input range of library fragments
be further prepared for sequencing. Therefore, quantitation must be precise. It’s also important to consider whether you’re quantitating productive library molecules – ones that will be (clustered and) sequenced – or if you’re simply quantitating total DNA or even total nucleic acid. Accurate quantitation of a library is essential for optimal sequencing outcomes, so choosing the right quantitation method may mean the difference between a successful run and a sub-optimal, or even failed, run, meaning
the library will then need to be adjusted and resequenced – an expensive and time-consuming proposition.
When choosing a quantitation method, there are many important considerations, including accuracy, throughput and cost. Several common methods are compared (Table 1) and discussed below.
| METHOD | EXAMPLE | BRIEF DESCRIPTION | BENEFITS/LIMITATIONS |
|---|---|---|---|
| Spectrophotometry (260/280) | NanoDrop™ | This method detects the absorption of UV light by the macromolecules in the sample. | ✓ Low cost, as most laboratories already have access to UV/vis spectrophotometers
✗ Not specific for DNA ✗ Results can be skewed by RNA or protein contamination ✗ Cannot determine fragment size |
| Fluorimetry | Qubit® | This method measures the enhanced fluorescence of a dye upon binding to DNA/ macromolecules. | ✓ Low cost, as most laboratories already have access to fluorimeters ✓ Can quantitate specifically dsDNA, ssDNA, RNA or protein ✗ Quantitates all nucleic acid present in sample, not just molecules to be sequenced ✗ Cannot determine fragment sizes |
| Electrophoretic | Bioanalyzer®, TapeStation®, Fragment Analyzer™ |
This method relies on capillary electrophoresis of DNA fragments for size estimation, as well as intercalating dyes for quantity determination. | ✓ Accurate determination of fragment size distribution
✗ Less reliable quantitation ✗ Requires expensive equipment |
| Quantitative PCR (qPCR) | NEBNext | This method measures fluorescence of a dye bound to dsDNA at each PCR cycle, quantitating relative to included standards. | ✓ Most accurate quantitation method
✗ More expensive ✗ Cannot determine fragment sizes |
What About Spectrophotometric Ratios and Flurometric Quantitation?
Due to their utility in multiple molecular biology applications, many labs already have spectrophotometers and fluorometers, and these enable relatively low cost quantitation.
For quantitating nucleic acid, spectrophotom- eters assess the amount of UV light absorbed by the sample at two wavelengths, 260 nm and 280 nm. A ratio of the absorbance values can then be used to determine whether or not the sample has contaminating proteins. The 260/280 ratio of a purified DNA sample should be between 1.7 and 1.9. Spectrophotometers are great at estimating the total amount and relative purity of nucleic acid in solution, but they can’t provide information about fragment size, and they can be confounded by an abundance of either RNA or protein in the sample.
Fluorometers, unlike UV-Vis spectrophotome- ters, rely on nucleic acid-specific dyes to assess the amount of nucleic acids in the sample. In this way, they avoid the pitfalls of spectropho- tometry, and can specifically quantitate dsDNA, ssDNA, RNA, or protein, depending on the dye used. However, they too are limited to gathering data about the entire complement of dsDNA or ssDNA in the sample, and not just molecules that will be sequenced.
It is generally recommended not to use only spectrophotometry, fluorometery, or even a combination of the two as your sole quantitation method before sample loading.
What can electrophoretic methods/ instruments tell me?
Electrophoretic instruments, such as the Agilent Bioanalyzer, TapeStation, and AATI Fragment Analyzer, provide valuable data in a variety of forms. The output of these instruments is a visualization based upon laser excitation of an intercalating dye during the sample’s passage through a chip matrix, and measurement of the time taken to travel through the matrix. The data can be formatted to look like the familiar band- ing pattern of gel electrophoresis or as a graph (a “trace”). On-chip electrophoresis enables faster, more standardized quantitation of nucleic acid samples than standard slab gel electrophoresis, with much smaller sample amounts.
Overall, electrophoretic instruments are excep- tionally useful tools for library quantitation, and they are a part of many laboratories’ NGS workflows. Electrophoretic methods are able to determine both the average library size and the size distribution of the library (important as a tight size range is generally more desirable than a broad size range). Still, electrophoresis-based quantitation of NGS libraries is not as accurate or consistent as qPCR-based methods, and is not specific for adaptor-ligated fragments. Additionally, electrophoresis-based methods are not sufficient for quantitating PCR-free library construction, as there is no PCR enrichment of adaptor-ligated molecules and they cannot discern between adaptor-ligated DNA molecules and unligated molecules.
qPCR: What’s in a name?
As the name implies, qPCR (or quantita- tive PCR) can provide an additional level of information about your library. Beyond simply reporting the total amount of DNA in your sample, qPCR-based library quantitation uses specific primers that hybridize to the adaptor sequences and, therefore, measures only mole- cules with adaptor sequences at both ends. This added specificity ensures that the fraction of the library loaded onto the sequencer contains the expected number of adaptor-ligated molecules. As described above, exact titration of adaptor-ligated DNA molecules is important for NGS as only molecules with an adaptor on each end can be successfully processed through the sequencing workflow. The quantity of the library is determined by comparing to a standard curve generated from DNA standards (known concen- trations of DNA of a known size), followed by a simple calculation to account for any difference in size between the library being measured and the DNA standards. Methods such as electro- phoretic analysis, described above, are useful for library size determination.
qPCR-based methods, which quantitate DNA sequences that are attached to adaptors, will also quantitate adaptor-dimers (e.g., two ligated adap- tors without any intervening library sequence). The presence of excessive adaptor-dimers in your library can skew your quantitation, but if this situation is suspected, running the sample on a Bioanalyzer or similar instrument will be infor- mative. Frequently, qPCR-based quantitation methods and electrophoretic methods are used in parallel, to determine both the quantity and quality of your library.
Specific details on the use of qPCR-based library quantitation are available in the product manual for the NEBNext Library Quant Kit for Illumina which can be downloaded at www.neb.com/ E7630.
The Big Picture
So, which method for library quantitation is right for you? Your answer will depend on a number of factors that are specific to your
situation, including your laboratory’s preferred DNA quantitation method, the tools you have available, your source material, and the size and scope of your experiment. No matter which platform you’ll be sequencing on, it is important to accurately determine the amount of sequence-ready DNA present. As we’ve described, accurate quantitation makes a meaningful differ- ence in the quality of the data you’ll create and the overall value of your experiment, by ensuring generation of optimal cluster densities and the equivalent representation of multiplexed libraries when pooling.
Using a qPCR-based approach, as we’ve just reviewed, ensures the most accurate quantitation, providing optimal conditions for Illumina’s sequencing chemistries. To make NGS library quantitation more accurate and reproducible, New England Biolabs® (NEB®) offers the NEBNext Library Quant Kit for Illumina. This qPCR-based kit is compatible with a broad range of library insert sizes and GC content, and has a user-friendly workflow.
| PRODUCT | NEB # | SIZE |
|---|---|---|
| NEBNext Library Quant Kit for Illumina | E7630S/L | 100/500 reactions |
Written by Elizabeth Young, Ph.D., Fiona Stewart, Ph.D. and Eileen Dimalanta, Ph.D.

