Advances in next generation sequencing: How researchers at NEB are working to improve our understanding of the genome, epigenome and transcriptome.
by Joanne Gibson, Ph.D. & Betsy Young, Ph.D.
Conducting innovative basic research has always been integral to New England Biolabs’ philosophy; our scientists have contributed to the advancement of science and modern molecular biology technologies for almost 50 years and, as a result, have authored or co-authored over 1,500 publications.
Basic research at NEB has led to the development of new technologies, streamlined workflows, and has helped facilitate a deeper understanding of the scientific questions we seek to answer. This empowers not only our scientists but also the scientists we serve - scientists working for scientists.
Advances in NGS are a major area of focus for NEB researchers. As scientific questions evolve, our research team works to fill technological gaps to enable more insightful genomic, epigenomic and transcriptomic analysis. Most recently, our scientists have applied their expertise in enzymology and NGS to address unmet needs in the analysis of genome-wide methylation, transcriptional start sites, full-length transcriptomes, chromatin accessibility, and much more.
This article gives an overview of some of the exciting sequencing technologies developed at NEB and discusses the applications they enable.
Methylation analysis
The most abundant form of epigenetic modification in the genomes of both prokaryotes and eukaryotes is methylation, which plays a role in gene regulation and cell differentiation. Methylome analysis has traditionally been restricted to sodium bisulfite treatment, which causes extreme damage, followed by short-read sequencing using platforms like Illumina®. With the recent introduction of NEBNext® nzymatic Methyl-seq (EM-seq™) (NEB #E7120) [1], it has become possible to analyze methylation across a genome without the challenges of conversion-induced DNA damage with sub-nanogram amounts of DNA.
Long-Read EM-seq (LR-EM-seq)
Long-read sequencing has steadily grown in popularity. Companies like Oxford Nanopore Technologies® (ONT) and Pacific Biosciences® (PacBio®) are facilitating ever-longer sequenc- ing read lengths. LR-EM-seq, developed in the Ettwiller lab, preserves the integrity of DNA using a highly effective enzyme-based conversion that minimizes damage [2].It allows long-range methylation profiling of 5mC and 5hmC within amplicons up to 5 kb using a long-read sequencing protocol. When applied to biologically relevant, differentially methylated genomic regions (DMR) with various methylation percentages and contexts, the result from LR-EM-seq is in accordance with previous studies.
Long-range phasing of methylated cytosines is essential for several applications, including studying DMRs of the genome, particularly where methylation status is a known disease biomarker. LR-EM-seq can also support haplotyping of methylation patterns and targeted methylation analysis.
This new sequencing technology is a comprehensive solution for analyzing 5mC and 5hmC methyl marks in various contexts (e.g., CpG, CHG, CHH). LR-EM-seq only requires a small amount of starting material – as low as a few ng of DNA. It uses the same analytical strategies developed for bisulfite sequencing.
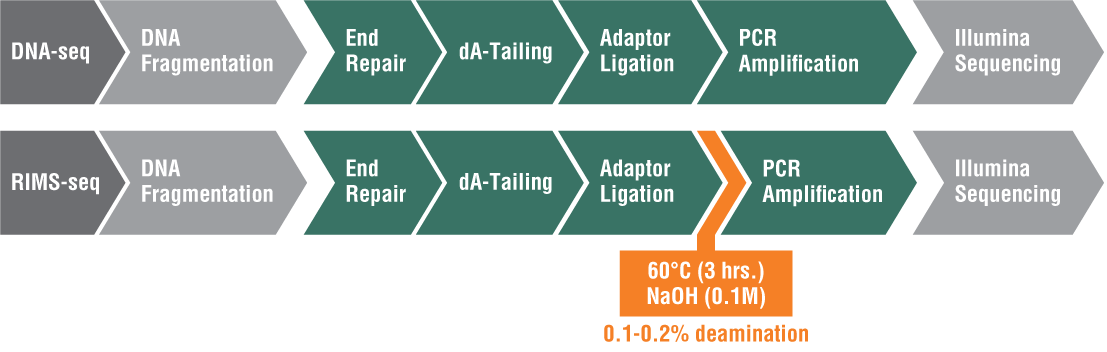
Rapid Identification of Methylase Specificity (RIMS-seq)
Also developed in the Ettwiller Lab, RIMS-seq seamlessly combines shotgun sequencing of bacterial genomes with 5mC methylase detection in a single experiment using Illumina sequencing[3]. There are three types of methylation in bacteria: 5mC (5-methylcytosine), 4mC (N4-methylcytosine), and m6A (N6-methyladenine). While PacBio single molecule real-time (SMRT®) sequencing can easily detect m6A and 4mC, it is more challenging to detect 5mC. This is because the polymerase stalls at the methylation site for a certain amount of time; however, the pause of the polymerase is much shorter for 5mC, which makes it more challenging to identify.
Inspired by the success of SMRT sequencing in revealing epigenetic landscapes, researchers in the Ettwiller lab modified the Illumina library preparation protocol and added an alkaline treatment incubation step. This treatment induces a level of deamination large enough to reveal 5mC methylation specificity commonly found in bacterial-RM systems while still producing sequence quality comparable to standard Illumina DNA sequencing. RIMS-seq can identify the 5mC methylome of mixed communities of unknown bacteria.
Because of the simplicity of this approach and its broad applicability, RIMS-seq has the potentialto replace standard DNA-seq for bacterial genome sequencing (Figure 1).
Identification of Transcription Start Sites (TSS)
Identifying transcription start sites (TSS) gives vital information relating to RNA transcripts, regulatory regions, promoters, and transcription factor binding sites in a sequence.
Cappable-seq
Developed in the Ettwiller and Schildkraut labs, Cappable-seq is a sensitive and robust method for directly enriching the 5′ end of primary transcripts from bacteria and microbiomes [4]. This method enables the determination of transcription start sites (TSS) at a single base resolution. Prokaryotes have a unique triphosphate at the beginning of the RNA transcript. One of the advantages of this technique is that it directly targets 5′ triphosphorylated RNA – the first nucleotide incorporated by the RNA polymerase upon initiation of transcription – in total RNA preparations. Cappable-seq uses this feature to capture the 5′ end of the molecule. The overwhelming majority of a total RNA sample is made up of processed RNA, such as ribosomal RNA, but by targeting 5′ triphosphorylated RNA, the rRNA population is reduced to just 3%, and the need to perform rRNA depletion beforehand is eliminated; therefore, it offers the ability to investigate the triphosphorylated RNA molecules that would otherwise be overwhelmed by processed RNA. This reduces the complexity of the transcriptome to a single quantifiable tag per transcript resulting in the ability to sequence the enriched 5′ triphosphorylated RNA population at a much deeper level at a lower cost, enabling the profiling of gene expression in a microbiome.
ReCappable-seq
The Ettwiller and Schildkraut labs built upon Cappable-seq with ReCappable-seq [5] to capture the TSS of non-RNA Polymerase II transcripts in addition to the TSS of 7-methyl G-capped transcripts derived from RNA Polymerase II. Therefore, ReCappable-seq overcomes the limitation of other methods that only determine RNA Polymerase II transcripts, which entirely exclude the TSS derived from eukaryotic RNA Polymerase I, RNA Polymerase III and mitochondrial RNA Polymerase that all produce uncapped non-coding RNA. To achieve this, they took advantage of the property of the yeast scavenger decapping enzyme (yDcpS) to convert capped RNA into di-phosphorylated RNA that can be “re-capped” by the Vaccinia Capping Enzyme, hence the name ReCappable-seq. Recappable-seq enriches both RNA Polymerase II- and non-RNA Polymerase II- derived transcripts and provides the ability to comprehensively evaluate both mRNA and non-capped primary transcripts all in one library, genome-wide, at single nucleotide resolution. It allows a unique opportunity to simultaneously interrogate the regulatory landscape of coding and non-coding RNA in biological processes and diseases.
Like Cappable-seq, ReCappable-seq produces sequencing libraries from total RNA without the need to deplete rRNA beforehand. Because it is species agnostic, it can be used with complex communities composed of both prokaryotic and eukaryotic organisms.
SMRT-Cappable-seq
This high-throughput technique was derived from Cappable-seq in the Ettwiller lab, but it differs in that it generates a snapshot of the full- length bacterial transcriptome at base resolution, whereas Cappable-seq identifies the TSS only [6].
To achieve this, the triphosphorylated 5′ ends of unfragmented transcripts are captured using an adapted Cappable-seq methodology, again removing the need to perform RNA depletion beforehand.
Because the transcripts do not need to be fragmented PacBio single-molecule long-read sequencing from TSS to the termination sites can be carried out. This is valuable when trying to gain information regarding bacterial operons, which are made up of a group of genes under the control of a common promoter – with short sequencing reads, much of the operon complexity is overlooked or hidden. Long-read sequencing keeps the information on the 5′ and 3′ ends of operons intact, facilitating their identification.
Additionally, SMRT-Cappable-seq can be used on complex microbiomes for which reference genomes are not readily available.
Analysis of chromatin accessibility
Within the nucleus, mammalian DNA is packaged as chromatin, along with essential proteins and RNA. The chromatin of the nuclear genome must be accessible to the transcriptional machinery to produce RNA for translation to cellular proteins. The dynamic nature of chromatin accessibility in cellular function is vital to gene expression and development.
Universal Nicking Enzyme-assisted Sequencing (UniNicE-seq ) and Nicking Enzyme-assisted Viewing and Sequencing (NiCE-view-seq)
Developed in the Pradhan lab, UniNicE-seq captures and reveals open chromatin sites (OCS) and transcription factor occupancy at single nucleotide resolution; it reveals the transcriptionally active genome [7,8].
UniNicE-seq utilizes a labeling mix containing a sequence-specific nicking enzyme (Nt.CviPII), DNA polymerase I and dNTPs (containing biotin- conjugated dATP, dCTP and 5-methyl-dCTP). This mix labels the open chromatin region in the nucleus; then, the DNA is extracted and sonicated/ digested to ~200 bp. This DNA undergoes librar preparation, and Streptavidin magnetic beads capture the biotinylated library components for further PCR library preparation and sequencing.
UniNicE-seq may be used with cell lines (formalin- fixed and unfixed), mammalian- or plant-tissue nuclei, frozen tissue sections, and formalin-fixed paraffin-embedded (FFPE) tissue sections. It can be used with high resolution and a broad range of cell number inputs (25-250,000). This technology is capable of automation and cell-to-library preparation in one tube.
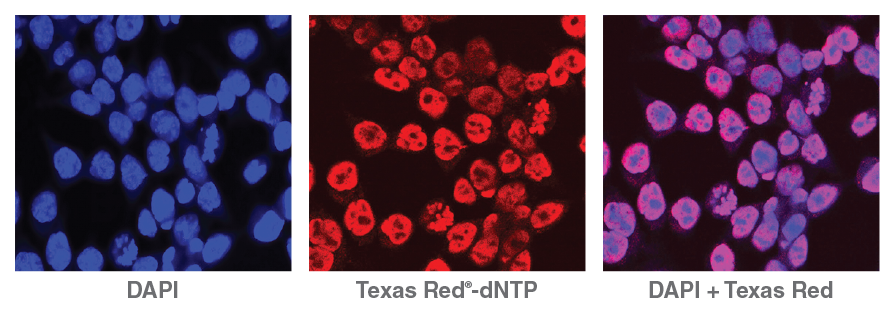
Lending an added dimension for analysis, NicE- viewSeq (Figure 2) includes a biotinylated dCTP mix and a Texas Red® tagged dATP mix that enables visualization of regions of accessible chromatin and subsequent sequencing for genome analysis, enabling pharmacological studies of chromatin- modifying drug efficacy [9].
Protect-seq
At the periphery of the metazoan nucleus, the nuclear chromatin becomes less accessible to transcriptional machinery and, in some cases, in direct apposition to the nuclear lamina (in what are known as lamina-associated domains, or LADs). Understanding which sequences tend to become arrayed in these LADs is an essential step toward understanding their functions.
Using a familiar technique in a new way, the Pradhan lab developed Protect-seq [10]. It relies on a cocktail of nucleases targeted at degrading and removing the open and accessible chromatin, as in NicE-seq, but with the goal of leaving the less-accessible, sonication-resistant LADs for sequencing. It is an efficient way to identify constitutive heterochromatin around the nuclear periphery. Protect-seq is a simple, easy-to-use, cost- and-time-effective method that does not require actively dividing cells, specialized equipment, or reagents. The entire protocol can be performed in a day.
Techniques that strive to answer similar questions require particular constructs, genome modifications, the establishment of cell lines, actively dividing cells or highly specific antibodies. In contrast, Protect-seq requires only an enzyme cocktail and nuclei from fixed cells or tissues.
This sequencing technique is compatible with short-read Illumina sequencing and long-read ONT sequencing, broadening its accessibility for researchers.
Analysis of DNA damage and modification
All living cells are exposed to DNA-damaging agents that are found exogenously, such as UV radiation, and endogenously, such as reactive oxygen species. These DNA-damaging agents can cause the formation of a wide variety of DNA lesions that can be mutagenic and cytotoxic to the cell. Cells have evolved several DNA repair pathways that recognize, remove and repair these DNA lesions. In higher eukaryotes, the formation of DNA lesions and faulty repair has been shown to cause cancer, neurological disorders and premature aging.
RAre DAmage and Repair sequencing (RADAR-seq)
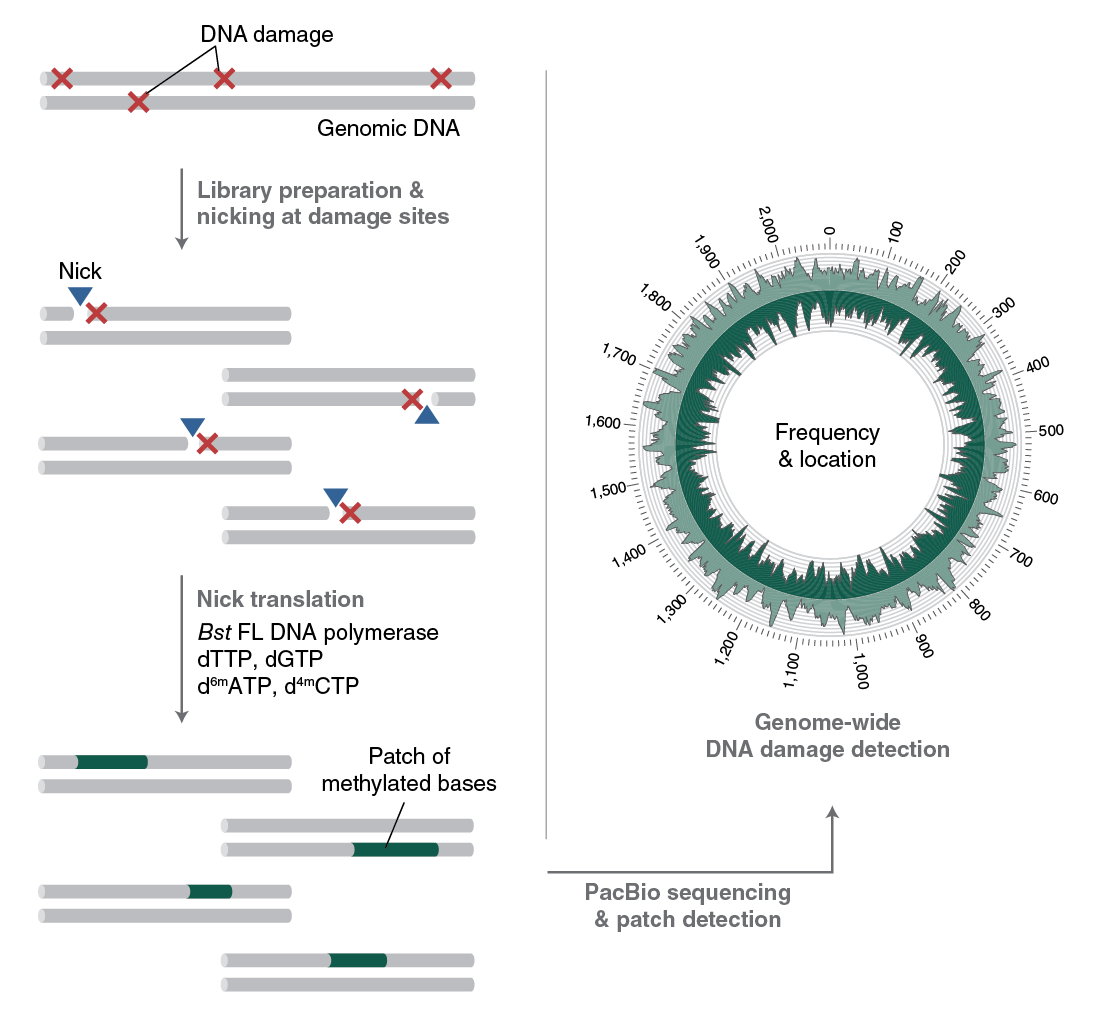
To understand the formation, persistence and repair of DNA lesions, NEB scientists have developed RADAR-seq [11,12]. This technique replaces a DNA lesion with a patch of modified bases that PacBio SMRT sequencing can detect. RADAR-seq can measure the frequency and map the locations of various rare DNA lesions genome-wide without of a specific DNA damaging agent, locate DNA damage hotspots across a genome, or locate the specific genomic site of any DNA nicking enzyme.
Several currently used DNA damage detection techniques employ short-read next generation sequencing methods that amplify damaged DNA; the drawback is that only the enriched regions with damaged, and not undamaged, DNA are sequenced. This gives a relative measure of DNA damage without information regarding absolute levels of damage in a genome. In contrast, RADAR-seq utilizes the PacBio long-read sequencing platform, which provides information on both damaged and undamaged DNA. In addition, most DNA damage detection techniques are tailored to locate a specific DNA lesion (i.e., ribonucleotides or cyclobutane pyrimidine dimers). RADAR-seq can locate any DNA lesion with an associated nicking enzyme or repair glycosylase. Furthermore, RADAR-seq can detect rare DNA damage, as low as one lesion per 1 Mb bases sequenced. Finally, RADAR-seq library construction is fast and can be completed in less than one day (Figure 3).
EcoWI-seq
This sequencing method was also developed in the Ettwiller lab [13] and can determine the pattern of phosphorothioate (PT) modifications in bacteria. PT modifications occur on the DNA sugar- phosphate backbone rather than on the nucleotide (a non-bridging oxygen is replaced by sulfur) and are maintained at a particular density in a genome. The PT modification is widespread in prokaryotes and is a horizontally transferred epigenetic system that makes the phosphorothioate oligonucleotide more resistant to degradation. The modification occurs on a small proportion of the EcoWI recognition sequences in the genome, and whether they occur stochastically or deterministically is an area of investigation.
The restriction enzyme EcoWI is a PT-dependent endonuclease with recognition sequence GAAC/ GTTC. EcoWI creates a double-stranded break only when a PT modification is present on both DNA strands. Subsequent sonication generates short sequences compatible with Illumina sequencing. The ability to detect PT modifications relies on the mapping pattern: fragments generated from sonication will map to the genome at random positions and provide sequencing information that can be used for genome assembly; fragments generated by EcoWI digestion map to fixed pattern ends, which provide the location of the modification at base resolution. This methodology is scalable and requires little starting material.
Enzyme discovery
The discovery and characterization of new enzymes aids the development of new technologies. Microbiomes are an untapped resource for discovering enzymes that can be harnessed for industrial purposes. Metagenomic, epigenomic, and transcriptomic pipelines are being used to rapidly discover novel enzymes.
Metagenomics Genome-Phenome Association (Meta-GPA)
The field of microbiome research has evolved rapidly and is a topic of great scientific and public interest. Nonetheless, microbiome studies are often limited to shotgun sequencing providing detailed descriptions of species composition and gene content, but direct links to function are missing. In other words, while we now understand who (species) are out there, it remains very difficult to understand what they are doing.
MetaGPA bridges this fundamental gap between genetic information and functional phenotype using next generation shotgun metagenomic sequencing [14]. MetaGPA is conceptually close to Genome-Wide Association Studies (GWAS), where control and case cohorts are compared to identify associated variants in the case cohorts. Likewise, metaGPA associates genetic data with phenotypic traits at the level of an entire microbiome. The association analysis can be done at the pathway, protein, or even single amino acid resolution level, pinpointing the residues within a protein domain that underlie a respective phenotype within a microbiome, irrespective of whether the organisms within that microbiome are known or culturable.
Because sequencing is conducted on environmental matter, there is no reference genome. Therefore, once the DNA is isolated and sequenced at a deep level, a de novo reference meta-genome must be created using a de novo assembler. The case and control genomes are plotted to reveal those with a high frequency in the test group. An enrichment score calculation associates the contigs with the likelihood of being a feature of the case group. These contigs are then analyzed for functional units.
Conclusion
At NEB, our scientists are passionate about developing sequencing technologies that can keep pace with their imaginations. As they continue developing new and exciting methodologies
for diverse applications to analyze the genome, epigenome and transcriptome, they also continue to foster NEB’s ongoing commitment to sharing knowledge and collaborating with the broader scientific community.
Learn more about the exciting research performed in the Ettwiller, Pradhan, Gardner and Schildkraut labs at www.neb.com/research.
References
- Technical note, NEBNext® Enzymatic Methyl-seq (EM-seq™)
- Sun, Z. et al. (2021) Genome Res., 31(2):291-300
- Baum, C. et al. (2021) Nucl. Acids Res., 8;49(19):e113
- Ettwiller, L. et al. (2016) BMC Genomics, 17:199
- Yan, B. et al. (2022) Genome Res., 32(1):162-174
- Yan B. et al. (2018) Nat. Commun., 9(1):3676
- Chin, H.G. et al. (2020) Clin. Epig., 12:143
- Vishnu, U.S. et al. (2021) Epig. & Chrom., 14(53)
- Estève, P-O. et al. (2020) J. Mol. Biol., 5304-5321
- Spracklin, G. and Pradhan, S. (2020) Nucl. Acids Res., 48(3)e16
- Zatopek, K.M. et al. (2019) DNA Repair, 36-44
- Zatopek, K.M. et al. (2022) Curr. Prot., 2:11
- Yang, W. et al. (2022) PLoS Genet., 18(9)
- Yang, W. et al. (2021) Elife, 8;10:e70021
View a PDF of this feature article

