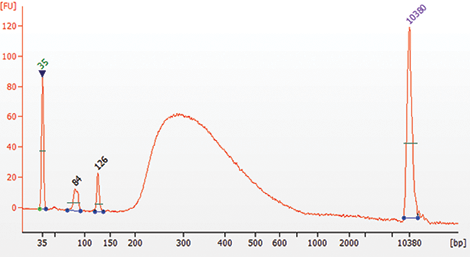
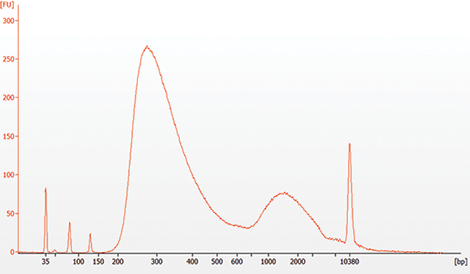
Appendix A
Note: These recommendations have been optimized using Universal Human Reference Total RNA. Other types of RNA may require different fragmentation times.
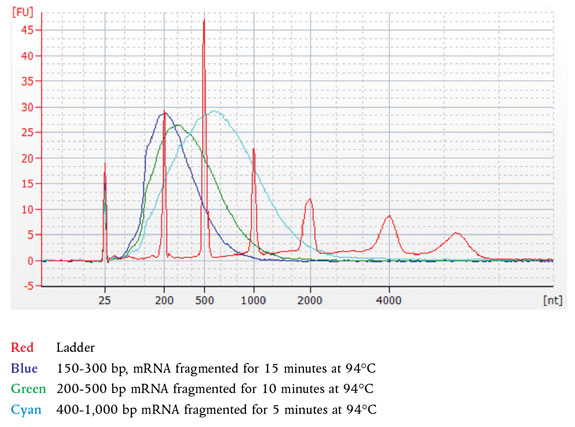
6.2. Size Selection of Adaptor Ligated DNA
Note: Size selection should be done after adaptor ligation and USER digestion.
 The size selection protocol is based on a starting volume of 96.5 μl. Size selection conditions were optimized with SPRIselect Beads and NEBNext Sample Purification Beads; however, AMPure XP Beads can be used following the same conditions. If using Ampure XP Beads, please allow the beads to warm to room temperature for at least 30 minutes before use.
The size selection protocol is based on a starting volume of 96.5 μl. Size selection conditions were optimized with SPRIselect Beads and NEBNext Sample Purification Beads; however, AMPure XP Beads can be used following the same conditions. If using Ampure XP Beads, please allow the beads to warm to room temperature for at least 30 minutes before use.
Please adjust recommended bead volumes for each target size according to Table 6.2. The protocol below is for libraries with a 300 bp insert size (420 bp final library size).
Table 6.2: Recommended size selection conditions for libraries with insert sizes larger than 300 bp.
Note: Size selection for < 100 ng total RNA input is not recommended.
| LIBRARY PARAMETER | APPROXIMATE INSERT SIZE | 300 bp | 400 bp | 450 bp |
| Approx. Final Library Size | 420 bp | 520 bp | 570 bp | |
| BEAD VOLUME TO BE ADDED (µl) | 1st Bead Selection |
25 |
20 |
15 |
| 2nd Bead Selection | 10 | 10 | 10 |
Note: Any differences in insert sizes between the Agilent Bioanalyzer and that obtained from paired end sequencing can be attributed to the higher clustering efficiency of smaller sized fragments.
6.2.1. Vortex SPRIselect Beads or NEBNext Sample Purification Beads to resuspend.
6.2.2. Add 25 μl of resuspended beads to the 96.5 μl ligation reaction. Mix well by pipetting up and down at least 10 times.
6.2.3. Incubate for 5 minutes at room temperature.
6.2.4. Place the tube on an appropriate magnetic rack to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic rack. After the solution is clear (about 5 minutes), carefully transfer the supernatant containing your DNA to a new tube (Caution: do not discard the supernatant). Discard the beads that contain the unwanted large fragments.
6.2.5. Add 10 μl resuspended beads to the supernatant, mix well and incubate for 5 minutes at room temperature.
6.2.6. Place the tube/plate on an appropriate magnetic rack to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic rack. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant that contains unwanted DNA. Be careful not to disturb the beads that contain the desired DNA targets (Caution: do not discard beads).
6.2.7. Add 200 μl of 100% freshly prepared ethanol to the tube while in the magnetic rack. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
6.2.8. Repeat Step 6.2.7 once.
6.2.9. Air dry the beads for up to 5 minutes while the tube/plate is on the magnetic rack with the lid open.
Caution: Do not over-dry the beads. This may result in lower recovery of DNA target. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack they are too dry.
6.2.10. Remove the tube/plate from the magnetic rack. Elute the DNA target from the beads by adding 17 μl of 0.1 X TE (provided) to the beads. Mix well on a vortex mixer or by pipetting up and down ten times. Quickly spin the tube and incubate for 2 minutes at room temperature.
6.2.11. Place the tube on a magnetic rack. After the solution is clear (about 5 minutes), transfer 15 μl to a new PCR tube for amplification.
6.3. PCR Enrichment of Size-selected Libraries
Note: Size-selected libraries require 2 additional PCR cycles due to loss during size selection steps compared to non-size-selected libraries.
Check and verify that the concentration of your oligos is 10 μM on the label. Use Option A for any NEBNext oligos kit where index primers are supplied in tubes. These kits have the forward and reverse primers supplied in separate tubes. Use Option B for any NEBNext oligos kit where index primers are supplied in a 96-well plate format. These kits have the forwardand reverse (i7 and i5) primers combined.
6.3.1. Set up the PCR reaction as described below based on the type of oligos (PCR primers) used.
6.3.1A Forward and Reverse
Primers Separate
| COMPONENT | VOLUME PER ONE LIBRARY |
|---|---|
| Adaptor Ligated DNA (Step 6.2.11) | 15 µl |
 (blue) NEBNext Ultra II Q5 Master Mix (blue) NEBNext Ultra II Q5 Master Mix |
25 µl |
| Universal PCR Primer/i5 Primer*, ** |
5 µl |
| Index (X) Primer /i7 Primer*, ** | 5 µl |
| Total Volume |
50 µl |
* NEBNext Oligos must be purchased separately from the library prep kit. Refer to the corresponding NEBNext Oligo kit manual for determining valid barcode combinations.
** Use only one i7 primer/ index primer per sample. Use only one i5 primer (or the universal primer for single index kits) per sample
6.3.1B Forward and Reverse Primers Combined
| COMPONENT | VOLUME PER ONE LIBRARY |
|---|---|
| Adaptor Ligated DNA (Step 6.2.11) | 15 µl |
| (blue) NEBNext Ultra II Q5 Master Mix |
25 µl |
| Index (X) Primer/i7 Primer Mix* | 10 µl |
| Total Volume |
50 µl |
*NEBNext Oligos must be purchased separately from the library prep kit. Refer to the corresponding NEBNext Oligo kit manual for determining valid barcode combinations.
6.3.2 Mix well by gently pipetting up and down 10 times. Quickly spin the tube in a microcentrifuge.
6.3.3. Place the tube in a thermocycler with the heated lid set to 105°C. Perform PCR amplification using the following PCR cycling conditions:
| CYCLE STEP |
TEMP | TIME |
CYCLES |
|---|---|---|---|
| Initial Denaturation | 98°C |
30 seconds | 1 |
| Denaturation | 98°C | 10 seconds | variable*, ** |
| Annealing/Extension | 65°C | 75 Seconds | variable*, ** |
| Final Extension | 65°C | 5 minutes | 1 |
| Hold | 4°C |
∞ |
* The number of PCR cycles should be adjusted based on RNA input. Size-selected libraries require additional 2 PCR cycles and should be adjusted accordingly. For example if a non-size selected library requires 8 PCR cycles, the size-selected library should be amplified for 10 cycles (8 + 2) after the size selection.
** It is important to limit the number of PCR cycles to avoid overamplification. If overamplification occurs, a second peak ~1,000 bp will appear on the Bioanalyzer trace (See Figure 7.2)
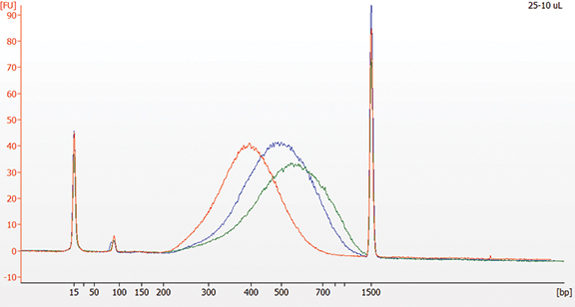
Table 6.3
| LIBRARY SAMPLE |
FRAGMENTATION TIME |
1ST BEAD SELECTION |
2ND BEAD SELECTION |
|---|---|---|---|
| Red | 10 minutes | 25 μl | 10 μl |
| Blue | 5 minutes | 20 μl | 10 μl |
| Green | 5 minutes | 15 μl | 10 μl |
For libraries with longer inserts (> 200 bp), remember to increase the incubation at 42°C from 15 to 50 minutes during the First Strand cDNA Synthesis reaction.