QC Check and Size Selection using AMPure XP Beads - NEBNext Multiplex Small RNA Sample Prep Set for Illumina (E7300)
- Purify the PCR amplified cDNA construct (100 μl) using a QIAQuick PCR
Purification Kit.
IMPORTANT: Before eluting the DNA from the column, centrifuge the column with the lid of the spin column open for 5 minutes at 13,200 rpm. Centrifugation with the lid open ensures that no ethanol remains during DNA elution. It is important to dry the spin column membrane of any residual ethanol that may interfere with the correct loading of the sample on the PAGE gel.
- Elute amplified DNA in 27.5 μl Nuclease-free Water.
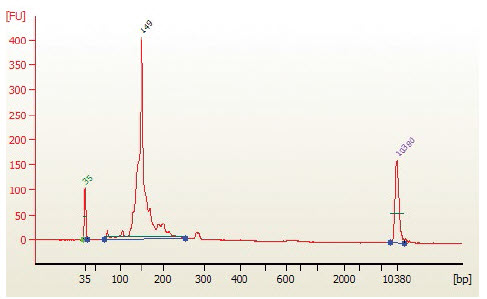
- Load 1 μl of the purified PCR reaction on the Bioanalyzer using a DNA 1000
chip according to the manufacturer's instructions (Figure 1).
- To the purified PCR reaction (25 μl), add 32.5 μl (1.3X) of resuspended
AMPure XP beads and mix well on a vortex mixer or by pipetting up and
down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Place the tube on an appropriate magnetic stand to separate beads from
supernatant. After the solution is clear (about 5 minutes), carefully transfer
the supernatant (57.5 μl) to a new tube (Caution: do not discard the
supernatant). Discard beads that contain the large DNA fragments.
- Add 92.5 μl (3.7X) of resuspended AMPure XP beads to the supernatant
(57.5 μl), mix well and incubate for 5 minutes at room temperature.
- Place the tube on an appropriate magnetic stand to separate beads from
supernatant. After the solution is clear (about 5 minutes), carefully remove
and discard the supernatant. Be careful not to disturb the beads that contain
DNA targets (Caution: do not discard beads).
- Add 200 μl of freshly prepared 80% ethanol to the tube while in the
magnetic stand. Incubate at room temperature for 30 seconds, and then
carefully remove and discard the supernatant.
- Repeat Step 9 once.
- Briefly spin the tube, and put the tube back in the magnetic stand.
- Completely remove the residual ethanol, and air dry beads for 10 minutes
while the tube is on the magnetic stand with lid open.
- Elute the DNA target from the beads with 15 μl nuclease-free water. Mix
well on a vortex mixer or by pipetting up and down, and put the tube in the
magnetic stand until the solution is clear.
- Transfer the supernatant to a clean PCR tube.
- Run 1 μl on the Bioanalyzer High Sensitivity chip. Check peak distribution and molarity of the small RNA library.
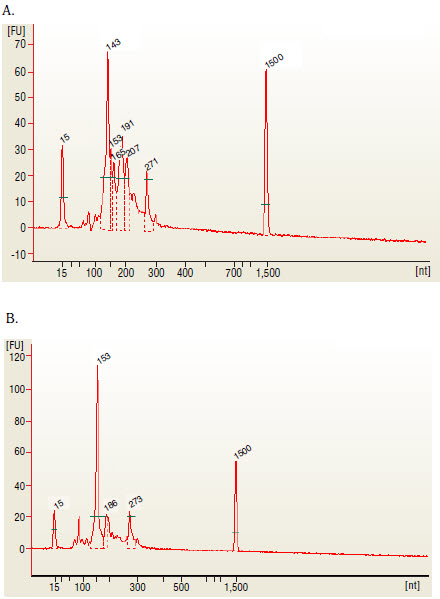
The 143 and 153 bp bands correspond to miRNAs and piRNAs, respectively. The bands on the Bionalyzer electropherograms resolve in sizes ~ 6-8 nucleotides larger than sizes observed on PAGE gels and can shift from sample to sample due to an incorrect identification of the marker by the bioanalyzer software. miRNA peak should be ~ 143-146 bp.