Protocol (E6040) for use with NEBNext Singleplex (#E7350) or Multiplex (#E7335, #E7500) Oligos for Illumina
Protocol
Starting Material: 1–5 μg of Fragmented DNA to 200 bpEnd Repair of Fragmented DNA
- Mix the following components in a sterile microfuge tube:
Fragmented DNA 1–85 μl
NEBNext End Repair Reaction Buffer (10X) 10 μl
NEBNext End Repair Enzyme Mix 5 μl
Sterile H2O variable
---------------------------------------------------------------------
Total volume 100 μl - Incubate in a thermal cycler for 30 minutes at 20°C.
- Vortex AMPure XP beads to resuspend.
- Add 160 μl (1.6X) of resuspended AMPure XP beads to the ligation reaction. Mix thoroughly on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of 80% freshly prepared ethanol to the tube/pcr plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target by adding 50 μl sterile water to the beads. Mix well on a vortex mixer or by pipetting up and down, and put the tube/pcr plate in the magnetic stand until the solution is clear.
- Without disturbing the bead pellet, carefully transfer 42 μl of the supernatant to a fresh, sterile microfuge tube.
dA-Tailing of End Repaired DNA
- Mix the following components in a sterile microfuge tube:
End Repaired, Blunt DNA 42 μl
NEBNext dA-Tailing Reaction Buffer (10X) 5 μl
Klenow Fragment (3´→ 5´ exo–) 3 μl
-----------------------------------------------------------
Total volume 50 μl - Incubate in a thermal cycler for 30 minutes at 37°C.
- Vortex AMPure XP beads to resuspend.
- Add 90 μl (1.8X) of resuspended AMPure XP beads to the ligation reaction. Mix thoroughly on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of 80% freshly prepared ethanol to the tube/pcr plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target by adding 30 μl sterile water to the beads. Mix well on a vortex mixer or by pipetting up and down, and put the tube/pcr plate in the magnetic stand until the solution is clear.
- Without disturbing the bead pellet, carefully transfer 25 μl of the supernatant to a fresh, sterile microfuge tube.
Adaptor Ligation of dA-Tailed DNA
- Mix the following components in a sterile microfuge tube:
dA-Tailed DNA 25 μl
Quick Ligation Reaction Buffer (5X) 10 μl
NEBNext Adaptor* 10 μl
Quick T4 DNA Ligase 5 μl
---------------------------------------------------------------------
Total volume 50 μl
* Adaptors can be purchased separately under NEB #E7335, #E7350, #E7500. - Incubate in a thermal cycler for 15 minutes at 20°C.
- Add 3 μl of USER™ enzyme mix by pipetting up and down, and incubate at 37°C for 15 minutes.
- Vortex AMPure XP beads to resuspend.
- Add 90 μl (1.8X) of resuspended AMPure XP beads to the ligation reaction (~53 μl). Mix thoroughly on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of 80% freshly prepared ethanol to the tube/pcr plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target by adding 105 μl sterile water to the beads for bead-based size selection as detailed In the next section, or at desired volume for size selection using E-Gel® (Life Technologies, Inc.) size select gels or standard 2% agarose gels. Mix well on a vortex mixer or by pipetting up and down, and put the tube/pcr plate in the magnetic stand until the solution is clear.
- Transfer 100 μl of supernatant (or desired volume) to a new tube/well, and proceed to bead based size selection.
Size Select Adaptor Ligated DNA Using AMPure XP Beads
| Insert Size |
150 bp |
200 bp |
250 bp |
300 bp |
400 bp |
500 bp |
700 bp |
| Total library size (insert + adaptor) |
270 bp |
320 bp |
370 bp | 420 bp | 530 bp |
660 bp |
820 bp |
| Bead: DNA ratio* 1st bead selection |
0.9X | 0.8X | 0.7X | 0.6X | 0.55X | 0.5X |
0.45X |
| Bead: DNA ratio* 2nd bead selection |
0.2X | 0.2X | 0.2X | 0.2X |
0.15X | 0.15X | 0.15X |
Table 1: Recommended Conditions for dual bead-based size selection.
Caution: the following size selection protocol is for libraries with 200 bp inserts only. For libraries with larger fragment inserts, please optimize bead: DNA ratio accordingly.
- Add 80 μl (0.8X) resuspended AMPure XP beads to 100 μl DNA solution. Mix well on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Place the tube/pcr plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully transfer the supernatant to a new tube/well (Caution: do not discard the supernatant). Discard beads that contain the large fragments.
- Add 20 μl (0.2X) resuspended AMPure XP beads to the supernatant, mix well and incubate for 5 minutes at room temperature.
- Put the tube/PCR plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets (Caution: do not discard beads).
- Add 200 μl of freshly prepared 80% ethanol to the tube/PCR plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 6 once.
- Air dry beads for 10 minutes while the tube/PCR plate Is on the magnetic stand with the lid open.
- Elute DNA target from the beads into 25 μl sterile water or 0.1X TE Buffer.
Mix well on a vortex mixer or by pipetting up and down, and put the tube/PCR
plate in the magnetic stand until the solution is clear.
Note: Be sure not to transfer any beads. Trace amounts of bead carry-over may affect the optimal performance of the polymerase used in the NEBNext High-Fidelity PCR Master Mix in the subsequent PCR step. - Without disturbing the bead pellet, carefully transfer 20 μl of the supernatant to a clean PCR tube and proceed to enrichment.
PCR Enrichment Adaptor Ligated DNA
- Mix the following components in a sterile microfuge
tube:
DNA 20 μl
Universal PCR Primer (25 μM) 2.5 μl
Index Primer (1)* (25 μM) 2.5 μl
NEBNext High-Fidelity 2X PCR Master Mix** 25 μl
---------------------------------------------------------------------
Total volume 50 μl
*Note: If you are using the NEBNext Multiplex Oligos for Illumina (Index Primers 1-12), for each reaction, only one of the 12 PCR primer Indices Is used during the PCR step.
** NEBNext High-Fidelity 2X PCR Master Mix will be replacing Phusion High-Fidelity PCR Master Mix. Both vials will be supplied for a limited time only.
- PCR cycling conditions
Cycle step Temp. Time Cycles Initial denaturation 98°C 30 sec 1 Denaturation
Annealing
Extension98°C
65°C
72°C10 sec
30 sec
30 sec4–8* Final extension
72°C
4°C5 min
hold1
*If library construction was performed with 5 μg of starting material, use 4 cycles of amplification. If starting material was 1μg, use 6-8 cycles of amplification. However, optimization of PCR cycle number may be required to avoid over-amplification.
- Vortex AMPure XP beads to resuspend.
- Add 50 μl (1X) of resuspended AMPure XP beads to the PCR reactions (~50 μl). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
- Incubate for 5 minutes at room temperature.
- Put the tube/ PCR plate on an appropriate magnetic stand to separate beads from supernatant. After the solution is clear (about 5 minutes), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain the DNA targets.
- Add 200 μl of freshly prepared 80% ethanol to the tube/PCR plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant.
- Repeat Step 5 once.
- Air dry the beads for 10 minutes while the tube/PCR plate is on the magnetic stand with the lid open.
- Elute DNA target from the beads into 30 μl of 0.1X TE. Mix well on a vortex mixer or by pipetting up and down, and put the tube/PCR plate in the magnetic stand until the solution is clear.
- Without disturbing the bead pellet, carefully transfer 25 μl of the supernatant to a clean LoBind® (Eppendorf AG) tube, and store at -20°C.
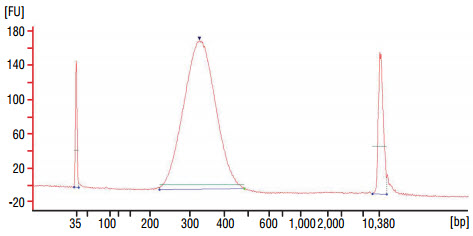
Alternatively, adaptor ligated DNA can be purified on one purification column. Purify DNA on one QIAQuick column and elute in 30 μl of 0.1X TE Buffer. - Dilute the library 20 fold with nuclease free water, and assess the library quality on a Bioanalyzer® (Agilent Technologies, Inc.) (high sensitivity chip). Check that the electropherogram shows a narrow distribution with a peak size approximately 300–320 bp.