Protocol for Control Reaction (E0554)
Protocol
A control mutagenesis reaction is included in this kit, allowing blue/white screening of mutagenesis efficiency. The Control SDM Plasmid is 6.7 kb and contains a premature stop-codon at amino acid #23 in the lacZα open-reading frame (1), as well as two BglII sites at positions #1948 and #2841. The Control SDM Primer Mix contains one primer that is 100% complementary to the Control SDM Plasmid and a second mutagenic primer. The mutagenic primer is designed to revert the aforementioned stop-codon to glutamine, thus allowing lacZα expression and blue colony formation upon transformation and plating on media containing X-gal and IPTG. The mutagenic primer also removes the BglII site at position #2841, allowing for restriction endonuclease screening of positive colonies. A BglII digestion reaction of the control plasmid will yield two DNA fragments of 893 bp and 5.8 kb. After a successful mutagenesis reaction, only a single DNA fragment of 6.7 kb will be present after BglII digestion.| CONTROL SDM PRIMER MIX | |
| Forward | AACCCTGGCGttAcCcAACTTAATCG |
| Reverse | TTCCCAGTCACGACGTTGTAAAA |
Step 1: Exponential Amplification (PCR)
For the control mutagenesis reaction, assemble PCR components as described in the following table:
| 25 μl RXN | FINAL CONC. | |
| Q5 Hot Start High-Fidelity 2X Master Mix | 12.5 μl | 1X |
| Control SDM Primer Mix | 1.25 μl | 0.5 μM |
| Control SDM Plasmid | 2 μl | 10 ng |
| Nuclease-free water | 9.25 μl |
Thermocycling Conditions for the Control PCR:
| STEP | TEMP | TIME |
| Initial Denaturation | 98°C | 30 seconds |
| 25 Cycles | 98°C | 10 seconds |
| 60°C* | 20 seconds | |
| 72°C | 1 min 45 sec | |
| Final Extension | 72°C | 2 minutes |
| Hold | 4–10°C |
For Step II (KLD reaction) and Step III (Transformation), follow the Q5® Site-Directed Mutagenesis Kit Protocol. Prior to plating, make a 40-fold dilution of cells in room temperature SOC (25 μl of cells and 975 μl of SOC) to avoid a lawn of colonies. For blue/white screening, spread cells on room temperature LB plates containing 100 μg/ml of ampicillin, 40 μg/ml X-gal and 50 μg/ml IPTG (stock solutions of X-gal and IPTG are made in dimethylformamide to 40 mg/ml and 50 mg/ml, respectively). Allow the plates to grow overnight at 37°C. For optimal color development, let plates sit at room temperature or 30°C for an additional 24 hours
Expected Results:
Exponential amplification with Q5 Hot Start High-Fidelity DNA Polymerase, combined with the intramolecular ligation reaction that precedes high-efficiency transformation, enables high numbers of transformants for simple mutagenesis
experiments (substitutions and deletions), and more complex strategies (insertions). In contrast, linear amplification methods typically generate fewer colonies in substitution experiments and are unable to perform large deletions
or insertions. Typical results from three different mutagenesis experiments with three different Q5 amplified site-directed templates are shown in the figure below. For substitutions and deletions, thousands of colonies were observed with
> 90% of the colonies having incorporated the desired mutation. An insertion of 18 nucleotides (6 amino acids for the addition of a 6X His tag) at the end of an open-reading frame still generated over 500 colonies. All isolates screened
for the proper insertion sequences were found to be correct. As with all PCR experiments, success rate is critically dependent on proper primer design. We highly recommend designing primers using NEBaseChanger, the NEB online primer design software.
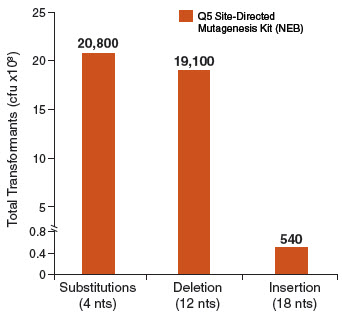
Typical results from a substitution reaction (4 nt) using the Control SDM Primer Mix, Control SDM Plasmid (6.7 kb) and NEB 5-alpha Competent E. coli (High Efficiency) Cells (NEB #C2987) are shown. For comparison, results from a 12 nt deletion experiment (5.8 kb plasmid) and an 18 nt insertion experiment (7.0 kb plasmid) are also shown. In all three cases, over 90% of the resultant colonies contained the desired mutation(s). Results are normalized to the total number of transformants if cells were not diluted prior to plating.

