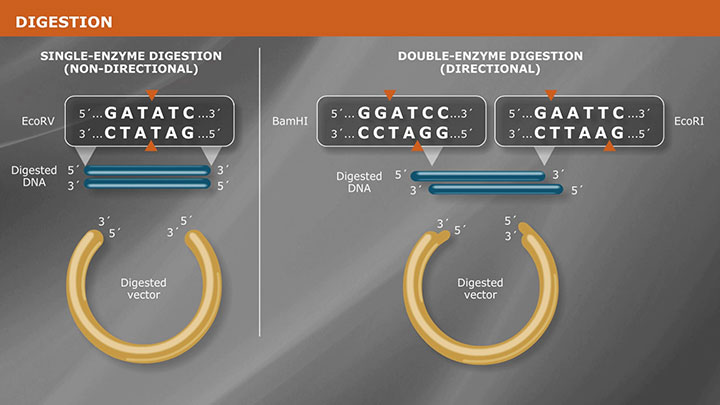
New to Cloning?
There are several methods and techniques available for cloning. Traditionally, cloning has utilized restriction enzymes to excise the DNA of interest, and to linearize a plasmid vector while creating compatible ends. After purification of the insert and vector, both are joined with the activity of a DNA ligase, and the newly-created recombinant vector is used to transform an E. coli host for propagation. PCR has also been used to generate both the vector and insert, which can be joined using a variety of techniques, ranging from standard DNA ligation or enzymatic joining using a recombinase or topoisomerase, to homologous recombination. More recently, DNA assembly techniques have been developed that utilize a mixture of enzymes to assemble DNA fragments in a single tube.
Learn more about the cloning workflow
Whether you are new to cloning, or having difficulties with an existing experiment, NEB offers a wide selection products, tools and resources that can help you be more efficient and successful with your experiments. To get started, choose the step in the cloning workflow below that you are interested in to find recommended products, videos, technical tips and more.









.

Selection Charts Brochures & Technical Guides Online Resources

Online Resources Brochures & Technical Guides



Online Resources OnlineTools



Selection Charts Online resources Brochures & Technical Guides Online Resources


Phosphorylation: Vectors and inserts digested with restriction enzymes contain the necessary terminal modifications (5´ phosphate and 3´ hydroxyl) for ligation to occur. Typical amplification does not use phosphorylated primers, and therefore the 5´ ends will need to be treated with a kinase, such as T4 Polynucleotide Kinase (NEB #M0201).
Dephosphorylation: If a vector is linearized by a single restriction enzyme, or has been cut with two enzymes with compatible ends, a phosphatase is needed to remove the 5´ phosphate, reducing intramolecular ligation and therefore background during transformation. It is important to note that if a vector is dephosphorylated, the insert must contain a 5´ phosphate for ligation to occur.
Blunting/End Repair: Blunting is a process by which the single-stranded overhang created by an RE digest is either “filled in”, by adding nucleotides on the complementary strand using the overhang as a template for polymerization, or by “chewing back” the overhang, using an exonuclease. Vectors and inserts are often blunted to allow non-compatible ends to be joined.
A-tailing: Tailing is an enzymatic method for adding a non-templated nucleotide to the 3´ end of a blunt, dsDNA molecule. This is typically done to prepare a T-vector, or to A-tail a PCR product produced by a high fidelity polymerase for use in TA cloning. TA cloning is a rapid method of cloning PCR products that utilizes stabilization of the single-base extension produced by Taq DNA Polymerase by the complementary T of the T-vector prior to ligation and transformation. It is important to note that this method is non-directional and the insert can go into the vector in both orientations.
Selection Charts Online Resources OnlineTools



Selection Charts Online Resources Online Tools


Selection Charts Online Resources Brochures & Technical Guides Online Tools




One or more of these products are covered by patents, trademarks and/or copyrights owned or controlled by New England Biolabs, Inc. For more information, please email us at gbd@neb.com. The use of these products may require you to obtain additional third party intellectual property rights for certain applications.
Featured Cloning Resources
ONLINE RESOURCES
| Getting Started with Molecular Cloning Explore simple tips to improve efficiency in your cloning experiment. |
|
| Troubleshooting Guide for Cloning Find common problems and solutions associated with cloning experiments. |
|
| Traditional Cloning Quick Guide Find quick tips for optimization of each step in the cloning workflow. |
ONLINE TOOLS
| NEBcloner® Find products, protocols and tips for each step of your traditional cloning experiment. |
|
| NEBioCalculator® For help with scientific calculations and conversions |
BROCHURES & TECHNICAL GUIDES
| Molecular Cloning Technical Guide Get help with product selection, protocols, tips for optimization and trouble-shooting. |