
High Molecular Weight (HMW) DNA Extraction – minimizing a bottleneck for emerging long read sequencing applications
Posted on Tuesday, February 16, 2021
By
Topic: What is Trending in Science
The number of applications that use data generated from sequencing long molecules of DNA, or HMW DNA, is expanding at breakneck speed. However, getting HMW DNA as an input material for these applications has been historically challenging and time consuming. Users of these emerging long read techniques are eager to find a solution to extract high-quality HMW DNA using a faster methodology, and one that is robust and user-friendly. Simplifying and speeding up this important step would enable researchers to carry out their long read downstream applications faster and more efficiently, producing a wealth of contextual genomic information with less effort.
Evolution of DNA sequencing technology
DNA sequencing has traditionally involved reading short strands of DNA; it is cost-effective and accurate. Sanger sequencing, while not strictly short read sequencing because it can produce individual reads of greater than 500 bp, was the mainstay of sequencing for 40 years and was crucial in producing the first reference sequence for the human genome. Next generation sequencing saw the introduction of technologies that enabled the sequencing of millions of short strands in parallel – providing the opportunity to sequence entire genomes at an unprecedented rate. In the third generation of sequencing technologies, the sequencing of long fragments of DNA as one single molecule removes the need to fragment and amplify DNA. The advantages of long read sequencing (LRS) are numerous: single-molecule sequencing without PCR amplification omits the GC bias that occurs during amplification; sequencing DNA in its native state enables detection of base modifications such as methylation and, working with a read length of 10kb+ makes genome assembly easier in a way that is analogous to putting together a puzzle with larger pieces. For example, if you think of the genome as a puzzle that you can either put together using 100 large pieces or 10,000 small pieces, the large pieces will be much easier to assemble, with less of a burden on bioinformatics. On the other hand, building a genome from short reads ultimately requires more sequencing depth to provide greater coverage.
Our current options for HMW DNA extraction are less than ideal
Anything larger than 50 kb is considered HMW DNA, but there is a rapidly developing need for DNA that is much larger than that – several hundred kilobases, even into the megabase range. One of the traditionally used and inexpensive methods for obtaining HMW DNA into the megabase size range is phenol/ chloroform extraction coupled with ethanol precipitation - a lengthy procedure working with hazardous chemicals. It is further hindered by the inevitable difficulty handling the HMW DNA - during the extraction process, it becomes an extremely viscous 'blob,' which is challenging to get back into solution before it can be used. The process of using phenol is also less than enjoyable – it requires working under a fume hood, has an unpleasant odor, and is toxic and therefore requires hazardous disposal.
There are newer methods that do not involve phenol/chloroform, such as magnetic bead-based approaches, and these also have pros and cons. There is the advantage of automation compatibility, which enables experiments to be scaled up for higher throughput. However, typical magnetic beads are tiny, and the very long DNA molecules bind and wrap around multiple tiny beads simultaneously. Subsequent centrifugation, vortexing or pipetting can cause the beads to separate and the HMW DNA to shear. So, although they offer improvements in scale, the size of DNA that can be extracted is usually around 100 kb.
A newer technology employing silica-coated magnetic discs was recently introduced and has gained some momentum. DNA into the megabase range can be isolated, and while the silica layer gives a large surface area for DNA binding, it binds very strongly to the disc giving a jelly-like layer of DNA, which is difficult to elute (lower yield) and subsequently difficult to dissolve (longer workflow).
How is NEB solving the HMW DNA extraction bottleneck?
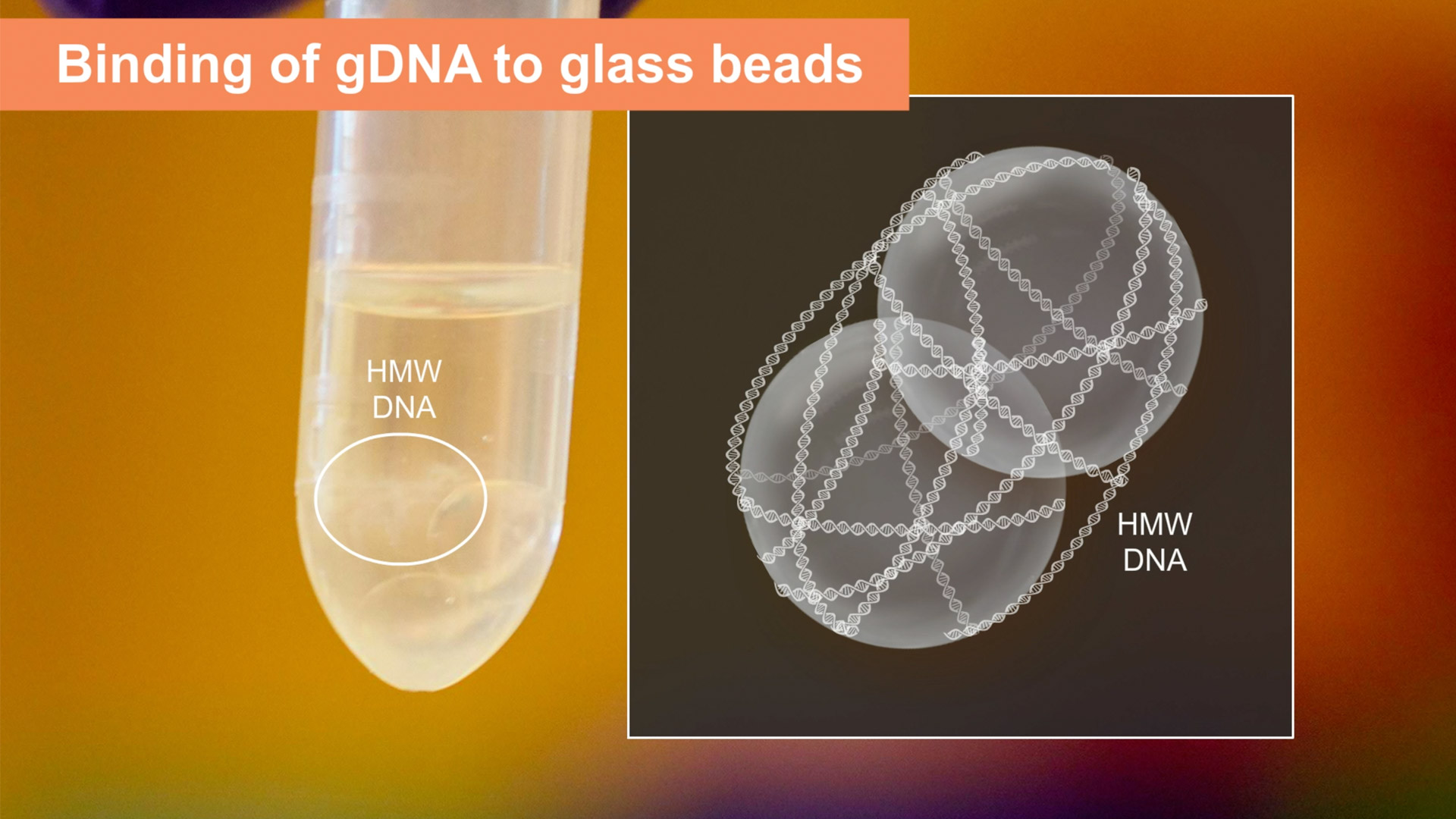
NEB researchers have developed a novel approach that yields intact, HMW DNA by utilizing glass beads. It addresses a couple of the shortcomings of other technologies. Firstly, the beads are large – 4mm in diameter – and smooth.

4mm glass beads for the extraction of HMW DNA
The lysis chemistry is optimized to inactivate nucleases and maintain the DNA integrity, and the lysis is carried out with agitation to control the DNA fragment size – faster agitation provides smaller DNA, and slower agitation provides maximally-long DNA. After lysis, the addition of isopropanol triggers the precipitation of a large web of DNA and the tube is inverted so that the DNA begins to stick to and wrap around the glass beads, kind of like the way cotton candy wraps around its stick. Because the long DNA molecules wind around the large beads, the DNA is extremely easy to elute – it basically slides off. The DNA is also captured in a relaxed and not compact form, allowing it to go back into solution with fairly minimal effort, which means that the DNA can often be used the same day it is extracted. Check out this application note where one user was able to shave 2-3 days off of his workflow.
The technology is available for processing cells & blood (NEB #T3050), or tissue (and bacteria and some other samples) (NEB #T3060), overcoming the sample limitations of some of the other extraction methodologies. The workflow is complete in 30 minutes for cells, 60 minutes for blood and 90 minutes for tissue and bacteria. This quick workflow also makes troubleshooting any issues with the downstream applications much easier. A future focus on expanding the repertoire of sample types is a high priority for NEB researchers, for example, optimizing lysis chemistry to overcome the binding inhibition caused by polysaccharides in plants, fungi and algae, and evaluating the breadth of microorganisms and invertebrates for which this approach can be used to isolate HMW DNA.
Overview of HMW DNA Extraction using the Monarch for Cells and Blood
What are researchers doing with long read sequences?
The need to extract intact, very HMW DNA quickly is essential so that researchers are not wasting precious time preparing the starting material for the ever-expanding array of technologies that are gleaning an abundance of genomic information. Some of the applications are based on sequencing these very long DNA molecules, and some do not necessarily require all of the information captured in a long sequencing read. So, what are researchers doing with this enormous amount of sequence information?
Currently, LRS enables an unprecedented rate of de novo assembly of a vast array of genomes - sequences for which there is no reference. Before the ability to perform long read sequencing, most reference sequences were assembled based on short read contigs, followed by the complicated task of assembling what ends up being a fragmented reference sequence. Using a long continuous stretch of DNA instead of shorter fragments gives the researcher much more contextual information around how the pieces fit together. For example, genome mapping is essentially making a map of chromosomes – assigning a specific gene to a region of a chromosome and determining the relative distances between and within genes on the same chromosome. Novel isoform identification is better served using long sequencing reads because it can cover an entire multi-exon gene. Overlapping exons and alternative splicing can make novel isoforms difficult to resolve using short reads. Structural variations such as large segmental translocations or inversions are easier to define and can be placed with greater confidence within the genome when using long, junction-spanning stretches of sequence. While short reads can identify changes at a single nucleotide level, or even small indels, the identification of larger sequence variations have proven difficult.
Some applications benefit from longer reads of DNA, even though not all the information captured in a long read is used. Genomic phasing, for example, refers to the ability to take into account the diploid nature of the human genome when looking at maternally and paternally derived sequences. Haplotype-based assemblies generated from longer reads of DNA give information regarding variants on two homologous copies of a chromosomal region and how this can impact the expression of the region's genes. The long read data generated from sequencing HMW DNA gives the ability to contextually associate different genome regions that may be variable in a meaningful way. This can give an understanding of allele-specific expression and genotype-phenotype associations. Additionally, when looking at base level modifications such as methylation, sequencing native molecules allows for their phasing with genetic variants, and this has applications in exploring epigenetic diversity. Optical mapping is another technique whereby all of the bases are not sequenced but a fluorophore is associated with high-resolution, ordered restriction maps to get long range information on how they fit into the chromosomal architecture.
Generally speaking, piecing genomes together with larger, continuous DNA sequences improves assembly contiguity and gives a greater understanding of the information contained within the genome than smaller fragments would. It is usually at a trade-off of the overall depth of sequencing – short read sequencing is cost-effective, and a greater depth of coverage is usually achieved when many reads are aligned. But, in reality, long and short read data is married in a way that, even though not looking as deeply, there is a depth of coverage generated by the short reads that complement the contiguity of the long read. So, in other words, the strengths of both sequencing methods are combined.
Genomes hold a wealth of information just waiting to be uncovered. The long read sequencing applications discussed here are just some of those emerging that have the potential to give exciting structural, epigenetic and phenotypic-related information. There is currently a lot of innovation and excitement in the HMW DNA extraction space. Reducing the extraction bottleneck is an essential step in paving the way to expedite the exciting new technologies that are unraveling the secrets of genomes.
Let us know in the comment section what samples you are working on and what techniques you are leveraging.
Check out our webinar where the Monarch® HMW DNA Extraction Kit product developer, Giron Koetsier, Ph.D., introduces the technology, walks through the workflow, and shares some long read sequencing data produced with purified HMW DNA.
NEB will not rent, sell or otherwise transfer your data to a third party for monetary consideration. See our Privacy Policy for details. View our Community Guidelines.
Don’t miss out on our latest NEBinspired blog releases!
- Sign up to receive our e-newsletter
- Download your favorite feed reader and subscribe to our RSS feed
Be a part of NEBinspired! Submit your idea to have it featured in our blog.


